Properties and Applications of Engineering Thermoplastics
1. Polyethylene Terephthalate (PET)
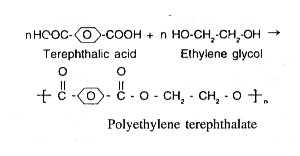
PET was discovered in 1941 by John Rox Dhirifield. It was commercialized in 1953. Engineering PET was introduced by Du pont in 1970's. In recent years, copolymers of PET have become increasingly important. PET is manufactured by condensation reaction between ethylene glycol (EG) with terephthalic acid (TPA) or dimethyl terephthalate (DMT). PET Polymerisation takes place in two stages. In the first stage, a prepolymer is formed by heating the reaction mixture and removing the volatile byproduct and excess glycol. In the second stage, heat and vacuum are employed to increase the molecular weight with concurrent removal of volatile products, such as water and glycol.
Characteristics
- Outstanding chemical resistance to organic and inorganic liquids.
- Water repellent
- Sterilizable by ethylene oxide and X-rays.
- Inherent good electrical property
- Ability to orient
- Good tensile strength
- Superior strength and stiffness
- Excellent dimensional stability
- Excellent melt strength with slower crystallization rate
- Good impact strength even at low temperature
- Excellent formability
- High tear strength
- Heat resistant
- Flame retardant
- Outstanding clarity
- High gloss

Applications
Appliances: Oven and appliance handles, coil forms for microwave oven transformers, small appliance structural frames, panels, chassis and housings and instrument covers
Automotive: Structural parts such as luggage racks and grills, functional housing such as windshield, wiper motor housings, fuel filters, blade supports and end bells, sensor, lamp sockets, relays, switches and solenoids.
Electrical/Electronics: Lamp sockets, coil forms for audio/video transformers, terminal blocks, electrical/electronic connectors and thrust washers.
Films: X-ray films, photographic films, magnetic tapes and printing sheets.
Furniture: Pedestal bases, seat pans, chair arms and casters.
Packaging: Carbonated soft drink containers, distilled spirits, toiletries, food products, mustard, pickle foods, peanut butter, spices, edible oil, cocktail mixers and syrups.
Miscellaneous: General purpose pump housings, mirror housings, three dimensional circuit boards and structural components in office automation equipments, computer housing and structural components, containers, trays, blisters and drinking cups.
Temperature Sensitivity
In the characterization of mechanical properties the previous three chapters have concentrated on the response of the material at ambient temperature, 23°C. While most structures spend most of their life near such a temperature, it is necessary to consider the response of the material over a range of temperatures that includes low temperatures as well as high. Civilian aircraft materials usually have to perform their duty in the temperature range - 50 to +80°C. For high-speed military aircraft skin temperatures up to 170°C may be encountered. High temperatures are also a feature of materials used in engines. There may also be adventitious exposure to very high temperatures in fires, tolerance of which may literally be a question of life or death. At the other end of the temperature scale is the possible use of composites for cryogenic storage of materials, including fuels such as liquid hydrogen. It is therefore necessary to consider the mechanical performance of thermoplastic composites from -270°C to +250°C, which latter temperature is usually considered to be the maximum continuous service temperature for polyetheretherketone in the form of an unfilled resin.
For structural composite materials the glass transition temperature represents a useful cut off point. For practical purposes a maximum working temperature of Tg minus 30OC has been suggested for load bearing structures. For polyetheretherketone this suggests a design temperature limitation of about 100°C. However, because PEEK is a semi-crystalline resin it is capable of useful service well above that temperature range in lightly loaded applications. The mechanical properties of PEEK resin have been characterized over the temperature range -80°C to +240°C for twelve decades of time. Since it is the resin whose properties are most sensitive to temperature, that characterization provides a vital starting point to the understanding of composite behaviour.
During the last five years there have been a number of programmes designed to develop new composite materials based on thermoplastic matrix resins with higher glass transition temperature, Tg. Typical of such developments are the melt preimpregnated product forms based on specifically designed matrices of the 'Victrex' family. These have included the semi-crystalline system APC (HTX), Tg 205°C, and the amorphous material APC (HTA). Tg 2600C. While semi-crystalline systems such as HTX (high temperature crystalline) and ITX (intermediate temperature crystalline) behave in a similar way to PEEK and retain some useful properties above Tg, the amorphous systems, HTA and ITA, which have the advantage of significantly higher glass transition temperature, must not be used above that temperature. Despite the considerable activity in developing such new systems, composites based on PEEK remain the most extensively characterized materials - in the open literature at least.
In the comparison of properties over a wide temperature range it is not always convenient to measure tensile properties directly. There can be difficulties in testing the required long specimens at very low temperatures, and at high temperatures significant problems of gripping the ends of the specimen are encountered. Flexural testing is a more versatile, if less precise, method from which comparative stiffness and strength can be deduced and, in this chapter, data from such testing, as well as from tensile and compression tests, are included. Where both flexure and tensile testing have been used, there is good qualitative agreement as to the influence of temperature on stiffness, but there may be quantitative differences in respect of strength measurements. Because flexure covers both shear and compression as well as extension, the flexural strength of axially aligned composites tends to be lower than the tensile strength, but transverse flexural strength is higher than transverse tension. For detailed results the reader is invited to refer to the publications cited, but, as a guide, the data in this chapter quote apparent properties relative to those at room temperature.
The Effect of Temperature on Stiffness
Table 1 collects typical reports of the stiffness of carbon fibre/PEEK composites relative to their stiffness at 23°C.
The only significant discrepancy in this table is the stiffness of the transverse specimens at high temperature. In his testing Leach used dynamic mechanical analysis and so had a short time scale for his tests. Hartness used tensile tests, and the stiffness may be presumed to be measured at much longer times. The results of Hartness are also based on 52% volume fraction, whereas Leach used 61%, so that matrix effects should be more significant. The difference between the results is consistent with creep mechanisms which are known to be significant near the Tg of PEEK. In respect of such creep behaviour, it is important to recognize that Ha reports that this response can be adequately predicted by a knowledge of the matrix behaviour, which has been thoroughly characterized by Jones.
Whereas creep is a problem for matrix dominated laminates at high temperature, the time dependent response of fibre dominated lay ups is the same at 120°C as it is at 23°C. Since good composite structure design would naturally arrange to have fibres oriented along the load path, we may conclude that the stiffness of such, correctly designed, laminates will not be affected by temperature in the range -268°C to +80°C. At 120°C there is likely to be a 5% reduction in composite stiffness as the matrix softens. At 180°C, 45°C above the glass transition temperature of the matrix,the stiffness of the composite is reduced by 10-15% and at 240°C, more than 100°C above Tg, the composite still retains 75% of its stiffness at room temperature.
2. The Influence of Temperature on Strength
Strength is a more complicated factor: it is necessary to consider^ compression as well as extension. There are also data available on the apparent short beam shear strength, a matrix dominated property. Typical results are collated in Table 2.
The results of Hartness are for composite based on 52% by volume of carbon fibre, and all other results are for 61% by volume carbon fibre: this difference may explain the fractionally lower results which he records. The resin sensitive properties -90° tension, short beam shear, ±45° tension, and uniaxial compression - fall off more rapidly than the fibre dominated properties, especially above the glass transition temperature. There is no significant loss of strength at very low temperature. Indeed there is evidence that the fibre dominated strength properties are significantly enhanced at temperatures close to absolute zero. As a first approximation we can say that, in the temperature range -268 to 120°C, the strength of carbon fibre reinforced PEEK composites based on 61% by volume of high strength carbon fibre is always at least 70% of the value measured at 23°C. At 180°C, 45°C above the glass transition temperature, this composite retains more than 40% of the strength properties that it has at room temperature.
The Applications of Thermoplastic Structural Composites
Structural materials, to which people must trust their lives, have to undergo a rigorous evaluation before they are put into service. Nowhere is this more true than in the aerospace sector, where the induction time for a new material to be extensively used is at least five, and typically ten, years. In the case of thermoplastic composite materials there is an added problem in respect of the development of appropriate manufacturing technology. The composite manufacturing industry has to make the transition from labour intensive hand lay up and protracted autoclave processes in order to exploit the high rate production potential of these new materials. In the evaluation stage, thermoplastic structural composites, and in particular preimpregnated carbon fibre reinforced polyetheretherketone, have demonstrated an excellent property profile, reproducibility, and durability: these designed materials are now beginning to take their place in serial production.
Carbon fibre/PEEK was designed as a material for aerospace. Innovative engineers have also demonstrated applications in the fields of prosthetic devices, marine technology, automotive engineering and specialized industrial machinery components.
1. Medical uses
One of the most successful operations in modern surgery is the replacement of hip joints. Bone is a living structure and, to be healthy, it needs stress in order to stimulate its growth and regeneration. Artificial hip joints made from stainless steel are stiff in comparison to bone, and this difference can cause the growth stimulating stresses to be reduced in the bone where the bone and prosthetic device join. The result of this loss of stimulus is a gradual deterioration of the bone and the possible need for remedial surgery. With composite materials there is the ability to tailor the anistropy of the fibres in such a manner that the stiffness of the prosthetic device can be designed to meet the needs of the bone1: the designer can tailor both the shape and the properties of his device. To be successful in such an application the material must at least be bio-inert, if not bio-compatible. Preliminary studies indicate that carbon fibre/PEEK meets these requirements. The toughness of this material is also of significance, since this allows it to be conveniently machined to shape without the danger of creating small fragments that might stimulate unwanted reaction from the body tissues.
Figure 1 shows a canine hip joint developed by Pfizer Howmedica. X-ray studies of the device one year after implantation have indicated a satisfactory condition of the surrounding bone.
As well as their use as implants, thermoplastic composite materials have potential as orthotic devices. De Lollis and Piancastelli note that high stiffness and strength per unit weight can enhance the confidence of the user. With thermoplastic composites there is also the potential for the structure to be conveniently tailored by post forming to meet individual requirements.
2. Satellites and Launch Vehicles
Nowhere are specific properties, stiffness, and strength divided by specific gravity, more highly prized than in space applications: Barnes notes that the cost of carrying a kilogram mass into orbit is of the order £5,000 to £10,000.
Launch vehicles themselves are heavy structures. Typical of the concepts for the next generation of such vehicles is the Horizontal Take Off and Landing vehicle HOTOL, described by Walmsley. The major structure is a fuel tank for liquid hydrogen, which must be capable of withstanding cryogenic shock without microcracking: and it appears that CF/PEEK is the only composite material which can do this. Evans confirms that the material does indeed have good resistance to permeation by hydrogen gas. In respect of fabrication such fuel tanks are cylindrical structures of very large size. Cylindrical structures are well suited to filament winding technology. Large filament windings are well suited to thermoplastic technology, because the infinite shelf life of the material means that the winding can be carried out over a long time, and because there is no requirement for large autoclaves in which to cure them. Walmsley considers carbon fibre reinforced PEEK as an appropriate material for such structures.
It is not just mass that contributes to launch costs. Volume is also a significant factor. Space station structures are inevitably bulky items. Spacecraft for deep space exploration are equally bulky and need not have the same structural characteristics as a vehicle designed to penetrate planetary atmospheres. Because thermoplastic composites can be shaped by heat and pressure, there is the potential to transfer the manufacturing process for bulky structures into space transporting there, in condensed form, only the materials and equipment necessary to carry out that task. On orbit fabrication should provide an important element of the next phase of mankind's exploration of space, and thermoplastic composites are well suited to play a role in that activity.
Earth launched satellite structures make extensive use of composite materials. Each year the United States Airforce TOPS (ThermOPlastics for Space) programme holds an annual workshop for companies working in this field. Typical of satellite structures being developed is an octagonal box structure, made from carbon fibre reinforced PEEK. That structure is remarkable for being built from prepregs based on both conventional high strength carbon fibre and ultra high modulus fibres; it also includes sub-components made from a hybridized staple fibre product form and conventional short fibre injection moulding. Such an approach to optimized structure design emphasizes the potential, optimally, to integrate the diversity of available thermoplastic systems.
In addition, Silverman and Jones note that carbon fibre reinforced PEEK composites have damping characteristics approximately three times as good as the current qualified grades of epoxy composites for this application. An important series of papers from the TRW Space and Technology Group, which includes an economic study demonstrating the advantages of thermoplastics, concludes that 'future spacecraft structural systems will be able to meet their projected flight requirements by using the superior performance and processing potential of thermoplastic materials over presently existing materials.
3. Aircraft Structures
Carbon fibre reinforced polyetheretherketone was first developed at a time of rapidly escalating fuel costs in order to enable commercial aircraft to operate more economically. The outstanding durability of structures made from such materials soon attracted military interest, and the United States Airforce has sponsored a series of annual workshops at which the progress of those materials to full scale application has been charted. Typical of the programmes that have been publicly disclosed is the development, by Lockheed, of a generic fighter forward fuselage structure and, by Westlands, of a tailplane for a helicopter. Extensive flight trials of demonstrator components have been in progress. Typical of such demonstrators is a carbon fibre/PEEK inspection panel on a Lynx helicopter that has been in service since 1983 and is showing no deterioration. The importance of inspection panels and hatches is that they are continually being removed from the aircraft so that ground technicians can maintain electronic and hydraulic systems. They are therefore subject to extensive handling and accidental damage. All reports from such trials have been uniformly encouraging.
One of the most significant demonstrator components has been the belly skin of a C-130 transport. During take-off and landing there is the possibility of runway debris being kicked up from the wheels, causing impact damage. The carbon fibre/PEEK belly skin of the C-130 is reported to have performed well during extended operation from 'unimproved' Alaskan runways, thereby confirming laboratory assessment of the toughness of the material under low temperature conditions. The underbody durability of carbon fibre/PEEK composites has also been verified by demonstration landing bay doors on military aircraft.
Because of the classified nature of military programmes, it is difficult to ascertain the extent to which thermoplastic composites are now being used. However, informed reflection indicates that, besides extensive demonstrator programmes, some aircraft primary structures made from thermoplastic composites have already entered production.
4. Marine applications
One of the longest serving demonstration components is on the leading edge of the rudder of a passenger carrying hovercraft. This component, installed by Westlands in 1983, has performed in exemplary fashion. As well as salt spray, normally a highly corrosive environment, there is the problem of impact resistance from sand and stones thrown up by the engines behind which the rudder is mounted. Those engines also provide a fatigue environment.
Besides the natural hazards of the ocean, fire and smoke are the most daunting enemies faced by those 'who go down to the sea in ships'. The fire resistance, and low smoke and toxic gas emission, of carbon fibre reinforced PEEK (Chapter 9), combined with stiffness, strength and durability, have attracted significant attention to this composite as a material system for naval construction. Most notable of such programmes is the vision for building submersibles from thermoplastic composites. As is the case with hydrogen fuel tanks, part of the attraction is to be able to carry out fabrication of very large structures over a long period of time without the need to consider the 'shelf life' of the material and without the requirement to build an autoclave big enough to 'cure' the structure. The ability to weld thermoplastic composite materials may also prove attractive to the shipbuilding industry. The relatively high cost of composite materials such as CF/PEEK in comparison to conventional material of marine construction may limit their use in that industry initially to specialized applications, but the property spectrum available from these materials may stimulate new design, which, in turn, could provoke the development of lower cost thermoplastic materials designed to the specific needs of the industry.
Applications of Differential Scanning Calorimetry and Polymer Characterization
Applications of DSC fall into a broad category of materials characterization, including thermal transitions in polymers: Tg, Tm; curing reactions and kinetics of thermosets, oxidation stability of lubricants and polymers; purity of pharmaceuticals and organics; specific heat capacity of materials and catalyst efficiency. The present article deals with evaluation of some of these parameters:
Specific Heat Capacity Measurement
Heat capacity is the amount of heat required to raise the temperature of a material. It is generally provided as specific heat capacity where heat capacity data is normalised to mass and temperature range. Thus, the units of specific heat capacity are joules per kilogram kelvin (or calories per gram degree celsius). Specific heat capacity information is tabulated at specific temperatures for it is a function which changes with temperature.
Differential Scanning Calorimetry, (DSC), reports heat flow as a function of temperature, is an excellent tool for the measurement of specific heat capacity. The measurement is made by heating a test specimen at a known and fixed rate. Once dynamic heating equilibrium of the specimen is obtained, the heat flow is recorded as a function of temperature. This heat flow, normalised to specimen mass and heating rate, is directly proportional to the specimen's specific heat capacity. In practice, two thermal experiments are required for each measurement. In the first, a baseline run is performed on only empty sample pan and lid. In the second run, the best specimen is enclosed in the pan and lid. The specific heat capacity information is derived form the difference between the two resulting thermal curves.
Heat flow calibration of the apparatus is also required. This is obtained by running baseline and experiment of traces for a material whose specific heat capacity is well known. Sapphire (a-alumina) is the calibration material of choice since it is readily available, and its specific heat capacity is known to five specific figures.
Calculations
The governing relationship for calculation of specific heat capacity is given in equation :

Cp (lit) is the reported literature value of specific heat capacity of the standard sample, while Cp(obs) is the experimentally observed value under the same conditions as are to be used in the determination of the unknown.
DSC Curing Kinetics
The Borchardt/ Daniels B/D DSC Kinetics Program permits the calculation of activatin energy (E), pre-exponential factor (Z), heat of reaction (H) and order of reaction (p) from a single DSC scan of a reactin exotherm.
Principle of Operation
The method assumes that the reactin follows order kinetics, i.e., obeys relationship:

The DSC exotherm is used to measure the two basic parameters required to solve these two equations:
- the reaction rate da/dt, obtained by dividing the peak height at temperature T by the total peak area; and
- the equation unreacted (1-a), obtained by measuring ratio of the partial area at temperature T to the toal peak area, to obtain a and then substracting this value from unity.
Applications
The B/D DSC kinetics method is applicable to determination of reaction rate information for such exothermic reactions as:
- Thermoset curing
- Chemical decomposition
- Thermoplastic polymerization
DSC Thermal Stability Kinetics
The ASTM E6988 DSC method measures exothermic maximum temperature variation with changes in linear programmed DSC heating rate. Several experiments are run at different heating rates (3 or more), between 1 and 20 K/min. and the temperature shift of the peak maximum is used to compute the kinetic constants.
The calculations make three assumptions:
- The peak maximum represents a point of constant conversion for each heating rate.
- The temperature dependence of the reaction rate constant obeys Arrhenius equation.
- The reaction is first order.
- Each of these assumptions can be expressed mathematically :
E=-2.16RXd(log b)/d(1/Tm)
Where: E = activation energy
R = gas constant (8.314 J/mole.K)
b = heating rate (K/min)
Tm = peak maximum temperature (K)
k = Z e(-E/RT)
Where: k = specific rate constant (min-1) at temperature T
Z = pre-exponential factor (min-1)
T = absolute temperature (K)
da/dt - K(l-a)
Where a = fractional conversion
t = time (min)
The solution to the first equation is obtained from measuring the slope of the log versus 1/Tm plot followed by computer literative calculation to define the value for the activatin energy (E). The pre-exponential factor (Z) is then calculated from the equation
Where T and b are obtained from a mid range heating rate.
Applications
This method is broadly applicable to exothermic reaction of these types:
- Decomposition of thermally unstable materials.
- Estimation of hazard potential of process reactions.
- Curing of thermosets.
- Polymerization of thermoplastics.
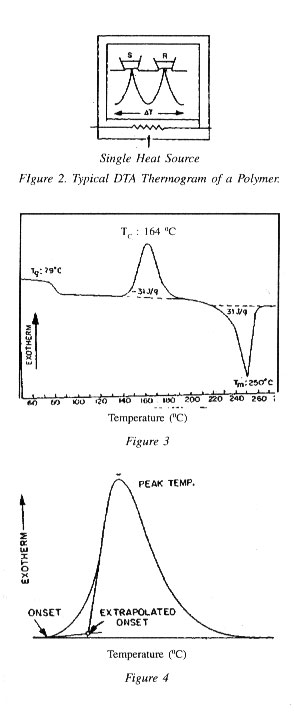
Degree of Crystallinity and Melting Point (Tm)
One of the most widely used application of DSC is the determination of Tm and degree of crystallinity of thermoplastic polymers. Since the DSC directly measures the amount of energy absorbed by the sample during melting the total area beneath the curve is directly proportional to the heat of fusion of the sample (DH). By ratio-ing the heat of fusion calculated with the DSC to the heat of fusion of the 100 : crystalline sample, the degree of the crystallinity of the polymer is readily calculated. For example the heat of fusion of a 100: crystalline sample of high density polyethylene is generally recognised as 286.186 J/g and if DH for a sample of HDPE is found as 185.9 J/g then % of crystallinity of the material would be 185.9/286.186 = 64.9% crystalline. Thus from a single scan, the melting point, heat of fusion and degree of crystallinity of a thermoplastic polymer can be determined.
Statement of the Problem
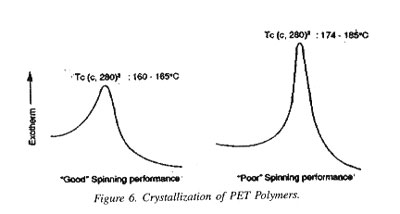
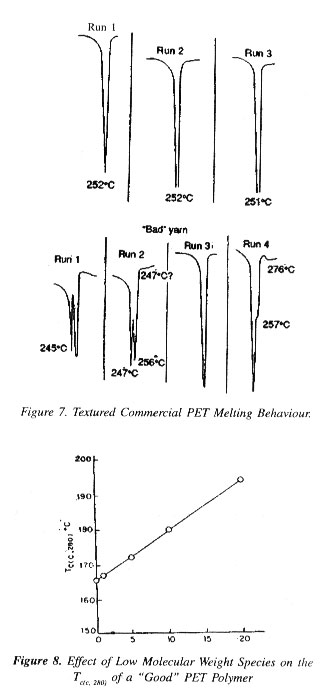
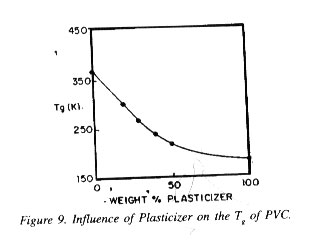
A.Polymer pellets manufactured under supposedly identical condit tions yield yarn of either good or poor spinning performance.
B. Non-thermal and some thermal methods failed to show differ ences characteristic to the spinning performance.
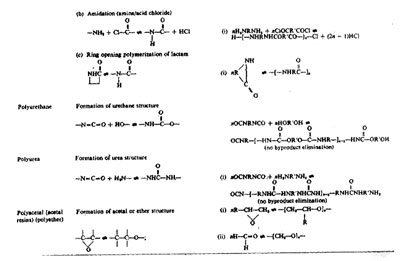
Condensation Polymerization or Step-Growth Polymerization
1. Functionlity Principal
Polymerization may be generally defined as intermolecular reaction between bifunctional or polyfunctional compounds (covering olefins and related compounds) avoiding formation of ring or cyclic structures and in a manner that makes the process functionally capable of proceeding to infinity. Functional groups or atoms, through disappearance of which polymerizations of different kinds take place are: reactive hydrogen atom (-H), hydroxyl group (-OH), carboxyl group (-COOH), amino group (-NH2), halogen atom (-Cl, -Br), isocyanate group (-NCO), aldehyde group (-CHO), double bond (C=C), etc.
According to the functionality principle and concept, for polymerization to take place, each of the reacting molecules must be at least bifunctional, giving average functionality for the system as 2. For reacting compounds containing two different kinds of functional groups, the average functionality is calculated on the basis of stoichiometric equivalence of the two kinds of functional groups. Thus, for reaction between a dicarboxylic acid and a trihydric alcohol, to strike stoichiometric equivalence of -COOH and -OH groups, the acid and the alcohol should be taken in a molar ratio 3:2, which would express the average functionality for the system as 12/5, i.e., 2.4. On this basis, any system consisting of a polyfunctional compound (functionality 3 or more) bearing a given kind of functional group (say, -OH) and a related monofunctional compound bearing a different kind of functional group (say, -COOH) would always give an average functionality less than 2. So, for any system, if one type of the reacting molecules is monofunctional, then the system would fail to produce polymer molecules. On consideration of functionality, a double bond (C=C) is bifunctional, as on thermal or photoactivation, the bond may open up to yield a diradical intermediate.
2. Types of Polymerization
Polymerization or growth of the polymer usually takes place by two basically different mechanisms. Depending on the mode of formation, one kind of polymerization is called condensation polymerization or step-growth polymerization, and the other kind is known as addition polymerization or chain-growth polymerization. The physicochemical aspects of condensation polymerization will be the subject matter of this chapter.
3. Basic Characteristics of Condensation or Step-Growth Polymerization
Condensation polymerization takes place under suitable conditions allowing bifunctional or polyfunctional molecules to condense intermolecularly in a manner that the growth of the molecule or chain extension takes place in controlled, distinguishable steps and the process is normally associated with the formation of condensed interunit chemical links and liberation of a small byproduct such as H2O, HCl, etc, at each step of reaction. Formation of a polyester by condensation polymerization or what may be alternatively termed as a polycondensation process between bifunctional monomers eliminating water as the byproduct of condensation may be cited as a typical example:
Formation of a Polyester

The condensation process beyond the first step need not necessarily involve reaction with one of the monomers. Condensation products of all the successive steps retain the bifunctional character, and each bifunctional species formed and remaining into the system at any point of time or extent of reaction is capable of reacting with any other bifunctional species including another of its own size or an unreacted monomer molecule, and the elimination of a molecule of H2O as a byproduct in each step remains a common feature. One more step of condensation results in the loss of one more bifunctional species and as a consequence two functional groups, one of each kind, are lost in each step. The formation of a long-chain molecule of degree of polymerization n, where n has a large value, may be expressed as follows, combining (2n-1) successive steps of condensation:
n HOROH+n HOOCR'COOHR
HO-[- ROOCR¢COO]n-H + (2n - 1) H2O (4)
The chain-growth process at any point of time may be represented by a general equation as follows:
SMn + ZMm ® ZMn+m + H2O (5)
where n and m may have any integral value between 1 and a. For the monomer, the value of n or m is 1.
For a hydroxy acid, the reaction would be written as:
n HORCOOH ® H-[-ORCO]n-OH + (n - 1) H2O (6)
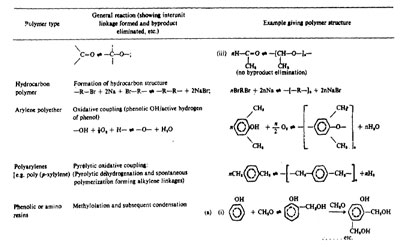
Examples of some step-growth polymerization are given in Table 1.

4. Relationship between Average functionality, Extent of Reaction and Degree of Polymerization
W.H. Carothers derived a general expression, relating average functionality (f), extent of reaction (p) and average degree of polymerization (), for polycondensation reactions carried out for a time period t. The expression is commonly known as Carothers' equation.
Let N0 be the total number of reacting molecules initially present giving an average functionality for the system as f. The related functional groups (say, - OH and - COOH) are also considered to be present in stoichiometric equivalence. Let N be the number of molecules present at time t when the extent of reaction is p.
Then, number of molecules lost during the process over the time period t is (N0 - N); for each molecule lost, the number of functional groups lost is 2 (one of each kind) and hence the total number of functional groups lost is 2(N0 - N) against the initial total number of N0 f functional groups.
Hence, the extent of reaction p=2(No-N)/NoF
or P = 2/f (1-N/No)
The average degree of polymerization, Xn, is defined as the average number of structural units per polymer molecule which can simply be expressed as:
combining Eqs. (7) and (8), one obtains
P=2/F(1-1/Xn)
Equation (9), known as Carothers' equation is very important for the understanding and control of the growth of polymers through poly-condensation reactions. The critical extent of reaction pc at the gel point (for Xn ® a) is given by:
5. Bifunctional System
For a bifunctional system, f = 2, equation (9) reduces to:
for p = 1,= = a, which means that for complete reaction, the average degree of polymerization will be infinity; but short of complete reaction, the degree of polymerization is always a finite quantity. Polymers obtained in a bifunctional system is, under all practical situations, linear and hence soluble and fusible. The upper theoretical limit in the degree of polymerization is infinity and this can be attained only in the hypothetical condition of completion of reaction between the functional groups (p = 1). For bifunctional systems, Eq. (10) may be rearranged to express Xn as
Xn=1/1-P
If the mean molecular weight of the repeat unit in the polymer molecules is Mo, then the average molecular weight Mn of the polymer system is expressed as
Mn=Xn.Mo=Mo/1-P
6. Polyfunctional System
A typical polyfunctional system in condensation polymerization is exemplified by the reaction between phthalic anhydride or phthalic acid and glycerol, i.e., by allowing a bifunctional acid to react with a trihydric alcohol. As discussed before, this system is characterized by an average functionality of 2.4.
For this system, Eq. (9) will assume the form:
P=2/2.4(1-1Xn)
We may now easily find out the critical extent of reaction, pc at which gelation or cross-linking would commence, i.e., at which point would first assume a value of infinity; the calculated value of pc, according to Eq. (9a), will be 2/2.4 = 0.83.
Thus, it is clearly seen that at the point of a theoretical extent of reaction p= 0.83, i.e., when the reaction is 83% complete, the polymer being formed in the polycondensation system gets cross-linked and, hence, turns into an insoluble, infusible gelled mass. Technically, such a condensation polymerization can not and should not be allowed to proceed beyond this point (83% completion), or otherwise the reaction mass would turn into an unusable product. In fact, it is advisable to arrest the reaction nearly 10-15% earlier in p to avoid damage of the reaction kettle and to collect a good quality product which would be useful and which could be easily manipulated in subsequent steps.
The usefulness of Carothers' Equation lies in the linking of p, f and in one expression, and in the fact that it can be used to predict gelation in polyfunctional systems. The limitation of the equation is that practically gelation starts even at earlier stages than predicted by the equation because of uncontrolled local conditions in the hot viscous reaction mass. Moreover, this equation is applicable to polycondensation reactions only, and it loses relevance in the context of addition or chain growth polymerization.
7. Molecular Weight Control: Quantitative Effect of Stoichiometric Imbalance on Maximum Attainable Molecular Weight
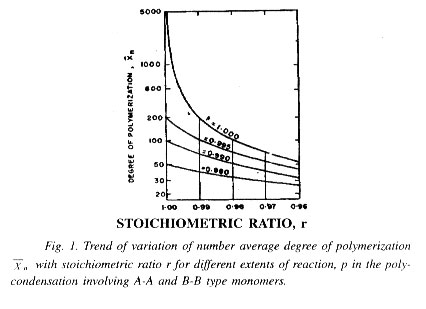
Condensation polymerization being an equilibrium step process and endothermic in nature in most cases, control of molecular weight can be readily achieved by allowing the reaction to die down at a desired point by withdrawing heat supply or by cooling and further, by allowing the byproduct of condensation to accumulate in the medium. But in any event, the intermediate product may not be adequately stable toward further heating under a favourable condition with respect to further chain extension and gain in molecular weight. Control of molecular weight can be more precisely attained by striking a calculated stoichiometric imbalance of the two types of reacting functional groups. This may be clearly illustrated considering a bifunctional system having monomers of the A-A and B-B types and giving the numbers of A and B functional groups as NA and NB respectively, such that (NA/NB) = r, where r is either equal to 1 or less than 1, i.e., B groups are present in the system in molar excess, if there is stoichiometric imbalance.
The total number of molecules initially present, No is then given by (NA + NB)/2, or NA(l + 1/r)/2. Let us now find out the total number of molecules N left into the system after a specific time period of reaction when extent of reaction for A groups is p, i.e., p is the fraction of A groups which have reacted. The fraction of B groups which have reacted by that time is then given by rp.
The total number of unreacted A and B groups are then expressed as (NA - NAP), i.e. NA (- p) and NB - NBrp, i.e., NB(I - rp) respectively. The total number of polymer chain ends is thus, given by [NA (I - p) + NB(l - rp)], i.e., [NA(l - p) + NA(l - rp)/r] The value of N, being one half this number, is given by N = NA[(1 - p) + (1 - rp)/r]/2.
Then,Xn=No/N= NA(1+1/r)/2/NA[1-P)+(1-rp)/r]/2
or Xn=1+r/1+r-2rp
and Lt p - 1,Xn=1+r/1-r
For equimolar proportions of A and B groups, i.e. for r = 1.0 = 1/(1 - p), which is the same as given by Eq. (11). The maximum theoretically attainable degree of polymerization for r = 1.0 is a. But for r ¹ 1.0, and with only, say one mole per cent excess of B groups taken into the system (by taking excess of the related bifunctional compound in calculated amount), and taking the help of equation (15), the maximum attainable becomes:
Xn=1+(100/101)/1-(100/101)=201
This consideration clearly establishes the importance of stoichiometric equivalence or balance of the reacting functional groups (A and B groups) for attaining high degrees of polymerization, or of introduction of stoichiometric imbalance in order to limit the maximum attainable degree of polymerization to a desired lower level. Fig. 2.1 shows how varies with variation in stoichiometric imbalance, i.e., variation in the value of r.
Considering a bifunctional system and Eqs. (11) and (15) applicable to it, it is clearly realized that the step-growth polymerization must always be carried out to very high conversions, at least to 98%, since a degree of polymerization ]50 is normally required for a polymer to be widely useful. Higher conversions and lesser deviation from stoichiometric balance are required for higher degrees of polymerization. The interdependence of on p and r is given by Eq. (14) and Fig. 1. Closer adherence to stoichiometric equivalence is required for obtaining higher degrees of polymerization. The effect of stoichiometric imbalance is much more pronounced in the very high conversion zone (]98%) than in the lower conversion zones.
8. Kinetics of Step-growth Polymerization
A powerful tool for a good understanding of any chemical reaction is the study of its kinetics. Kinetic analysis of a reaction helps in the understanding of the mechanism of the reaction.
Principle of Equal Reactivity of Functional Groups
Equation (5) describes a characteristic feature of step polymerization. Any two species in the reaction mixture can react with each other. This feature alone complicates kinetic analysis of the overall process, as it involves innumerable separate reactions. However, the kinetic analysis is largely simplified if it is assumed that the reactivities of both functional groups of a bifunctional monomer, e.g., both the carboxyl groups of a dicarboxylic acid, are just the same and that the reactivity of a functional group is independent of the size of the molecule to which it is attached. This means that the reactivity of the specific functional group is independent of the values of n and m. Fortunately, this assumption is more or less valid for all monomers and all larger species that are involved in polycondensation.
The independence of the reactivity of a specific functional group on molecular size is also generally substantiated by the closeness of the rate constants for reactions of a homologous series of compounds (fatty acid) differing from each other only in molecular (weight) size involving esterification with a specific alcohol (ethyl alcohol) in each case.
mineral acid
H(CH2)n-COOH + C2H5OH ---® H(CH2)n-COOC2H5 + H2O
catalyst (HCl)
Table 2, however, shows a decrease in reactivity with increase in molecular size (n) over the very (initial) low range of n and the rate constant of the esterification reaction levels off to a limiting (constant) value for n ³ 3 and maintains constancy and independence of molecular size. This feature of equal reactivity may be generalized and considered applicable to polymerization reactions as well. In the latter case, some complications not encountered in the study of simple compounds may arise, but they may be readily understood and accounted for when considered under proper perspective, i.e., equivalent (molar) concentrations of reacting functional groups, solubility, dilution level, etc. An end (functional) group comprising a terminal reactive centre of a polymer (chain) molecule has a much greater mobility than that of the polymer molecule as a whole. The functional group at the free end would have good degree of mobility in view of the rearrangements that would normally occur in closeby segments of the chain molecule. The collision rate of such reactive functional groups with related functional groups in the neighbourhood will be much the same as for small molecules.
Table 2. Esterification Rate Constants, k at 25°C for Homologous Series
Molecular size AB nkÃ-104kÃ-104 122.1_ 215.36.0 37.58.7 47.58.4 57.47.8 6-7.3 87.5 97.4 117.6 157.7 177.7
All k values are in unit of (gram equivalent/litre)-1, sec-1. A-Esterification between monocarboxylic acids, H(CH2)nCOOH and C2H4OH.B-Esterification between dicarboxylic acids (CH2)n(COOH)2 and C5H5OH.
In polymer systems, the medium viscosity, either in melt condition or in solution, is usually much higher than in systems containing only small non-polymeric molecules. Further, the polymer molecules may become insoluble beyond a critical chain length or molecular weight and the low molecular weight byproduct molecules would tend to slowly accumulate in the system causing the equilibrium to shift in favour of the reactants [Eq. (5)]. All these physical factors may affect the polymerization rate quite measurably. The rate at which independent functional groups react to produce chain extension through establishment of a fresh condensed interunit linkage is dependent on three different rate processes: (i) the rate at which the groups diffuse into a cage, (ii) the rate at which they diffuse apart, and (iii) the rate at which the reactive groups trapped in the cage react to produce the new condensed interunit linkage. Thus, in a given time interval long enough to allow diffusion of a pair of reacting groups, the number of related functional groups with which a given functional group may undergo collisions will be less for a group linked to a polymer molecule than for one linked to a small molecule. But what is more significant is that the overall frequencies of collision in either case are much the same. A pair of groups with a lower diffusion rate will undergo more total collisions before diffusing apart. The net outcome of these considerations points to the important conclusion that the reactivity of a functional group is independent of the size of the molecule to which it is linked.
Rate of Step-growth Polymerization
(a) Self-catalyzed Polyesterification. With the idea of equal reactivity of specific functional groups independent of molecular size, the consideration of rate of step-growth polymerization becomes easy. The formation of a polyester from a dicarboxylic acid and a diol may again be taken as an example, for simplicity. This reaction is prone to catalysis by mineral acids. But in the absence of an added mineral acid such as H2SO4 or HCl, the reactant carboxylic acid itself acts as a catalyst; for each step of reaction between a -COOH group
-COOH -COOH + HO- ----® - COO - + H2O (catalyst)
and an alcohol (-OH) group, a second COOH group from the reactants acts as a catalyst. The rate of the reaction can be measured by measuring the rate of disappearance of the carboxyl groups:
Here, k is the rate constant of the reaction given by equation (16). Considering equimolar presence of carboxyl and hydroxyl groups, and c giving the concentration of each group at a given point,
or on integration, Eq. (2.19) gives:
If co is the initial concentration of each kind of functional group and p is the extent of reaction, i.e., fraction of a given functional group that has reacted at time t, then
c = c0 (l - p)
By combining Eqs. (21) and (20), we get
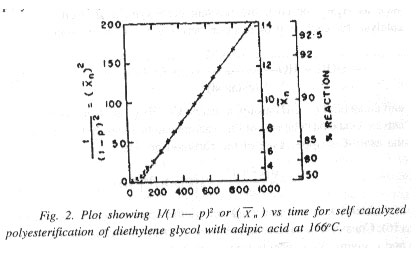
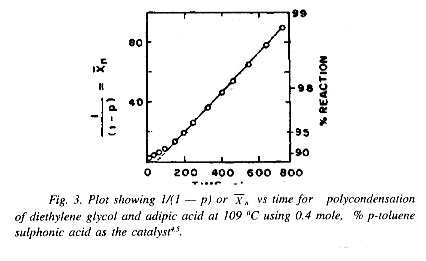
Equation (22) prescribes linearity of the plot of 1/(1 - p)2 vs time. In the case of self catalyzed polycondensation (polyesterification) of diethylene glycol and adipic acid at 166oC, as studied by Flory, the linearity is found to be strictly followed only over the range of nearly 80-94%. Below or above this range of extent of reaction, the experimental points deviated from the third order plot. Equation (22) actually shows a linear relationship between square of the average degree of polymerization, ()2 with time of polycondensation.
It may be mentioned here that the initial nonlinearity is not limited to polyesterification; the initial deviation from the third order linear plot is characteristic of esterifications in general.
Over the initial 50-80% conversion range, a very significant change takes place in the reaction medium, although the average growth in size of the molecules remains far too low. A large change in the polarity of the reaction mixture, however, takes place due to the disappearance of a large fraction of the polar carboxyl groups resulting in the formation of less polar ester linkages. This definite change in polar nature of the reaction medium has a major role to play in causing the deviation in question. But the exact manner or mechanism in which the rate of esterification is affected as a consequence of this polarity change is not very well understood.
The deviation from linearity in the high conversion region (]94%), however, arises as a consequence of other factors including loss of reactants through degradation or volatilization, or even cyclization and other side reactions. Such losses of reactants are likely to be of much less importance in the initial stages. Difficulty in removal of the byproduct of condensation (water) from the reaction medium due to the very high viscosity it assumes in the later stages is also an important contributing factor in decreasing the reaction rate with increasing extent of reaction beyond 94% conversion.
(b) Polyesterificationi Using Strong. Acids as Catalysts. The direct or self-catalyzed polyesterification is slow and sluggish, considering slow increase in molecular weight or average degree of polymerization and this is understood to be a consequence of the third order kinetics. A better result and a faster rate of chain growth is, however, achieved by adding a small amount of a strong acid such as sulphuric acid, p-toluenesulphonic acid, etc. as a catalyst which remains undepleted in the system with progress of the reaction. The rate of disappearance of COOH groups can then be expressed as
For stoichiometric equivalence of the two functional groups and taking c as the concentration of either group the equation (23) may be written as
where k' is the overall rate constant. On integration, and making use of Eq. (21), the following expressions, relating extent of reaction p or degree or polymerization with time of reaction, t, are obtained:
c0k¢t = - constant or c0k¢t = n - constant
A plot of 1/(1 - p), i.e. , vs time, would be linear according to Eqs. (25) and (26). Data relating to polymerization of diethylene glycol and adipic acid catalyzed by p-toluenesulphonic acid as observed by Flory4 are shown in Fig. 3. The plot shows the experimental variation of , the average degree of polymerization with time of polycondensation at a given temperature. Comparison of Figs. 2 and 3 clearly shows that the polyesterification process is much more readily and economically accomplished when an external strong acid catalyst is used. Not only the linearity of vs time plot is very closely maintained, particularly in the more important high conversion zone, but also the rate of increase of with time of reaction is much higher. The second order kinetics find good experimental support up to nearly 98.9% conversion corresponding to value of approximately 90 or a molecular weight of nearly 10,000. This approach of kinetic analysis, however, shows that the reactivities of the carboxyl and hydroxyl groups remain practically unchanged despite growth of the chain molecules to a fairly big average molecular size and a big increase in medium viscosity as a consequence.
Step-growth polymerization other than polyesterification may proceed at desired rates in presence of external acidic or basic catalysts or as uncatalyzed reactions. Irrespective of the approach and situation prevailing, the basic and overall kinetics will be much the same as detailed above for polyesterification. For stoichiometric proportions of the two types of reacting functional groups, say A and B groups, Eq. (25) would be useful to reasonably describe and analyze the kinetics in the respective systems. Whether or not the reacting monomers are of the A-A and B-B types or simply of the A-B types, the observed kinetics will be the same.
Copolymerization and Techniques of Polymerization
1. Concept of Copolymerization
A polymer whose chain molecules are composed of more than one kind of repeating chemical units is commonly called a copolymer. A simple case of a step-growth copolymer is the linear unsaturated polyester formed by inter-molecular condensation of a mixture of phthalic acid (anhydride) (PA) and maleic acid (anhydride) (MA) with a given diol, say ethylene glycol (EG) resulting in what may be termed as a copolyester with at least two distinctly identifiable repeating units in the chain molecules:
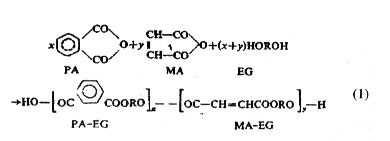
Equation (1), showing formation of a copolyester by a polycondensation process clearly identifies two types of repeating chemical units, (PA-EG) and (MA-EG), though in a very oversimplified manner. In the actual copolymer the two types of distinguishable chemical units would normally appear at random along the chain, and the overall composition would depend on the relative values of x and y. Similarly, one can prepare step-growth copolymers with different interunit chemical linkages by proper selection of combinations of different bifunctional monomeric species bearing, -OH, -COOH, -NH2, -NCO or other functional groups, and using two, three or all the functional species simultaneously or in a phased manner in the presence or absence of a related polyfunctional and monofunctional species during the synthesis. However, polycondensation leading to formation of copolymers from this kind of complicated mixtures of monomeric species is of little or rare practical importance or relevance. More relevant step-growth copolymerization systems involve formation of copolymer structures with a specific interunit linkage such as amide (-CONH), ester (-COO-) urethane (-NHCOO-), etc., following the approach as in Eq. (1). It may also involve chain extension and/or cross-linking reaction introducing similar or different kinds of interunit linkages, using low molecular weight linear or branched polymers and selected bifunctional monomeric species. Examples of this kind of step-growth copolymers are found in the making of polyester urethanes or polyether urethanes, using low molecular weight performed linear polyesters or polyethers with hydroxyl end groups and allowing them to further react with each other through diisocyanates leading to chain extension and producing useful elastomeric products commonly known as
An altogether different kind of copolymer is obtained by polymerization of two or more vinyl and related monomers, all taken together in a reaction vessel. The copolymer molecules contain segments of all the monomers used in their structure, usually in a calculable proportion depending on many factors including the feed monomer composition, the chain-growth mechanism and the inherent reactivities of the monomers with a given chain carrying species.
The number of reactions to be necessarily considered to describe the copolymerization of more than one monomer increases geometrically with increase in the number of participating monomers, giving increasingly varied and complicated structures of the copolymer molecules at the same time. A case of a binary copolymerization involving the two monomers Mî and M2 may be simply represented as:
nM1 + mM2 ~~~~ (M1)n-(M2)m ~~~~
monomer mixture copolymer
The structure of the vinyl copolymer shown in Eq. (4) is far too simplified, however; the two monomer units appear in the copolymer structure in a random sequence in general.
2. Binary Copolymerization of Vinyl Monomers by Free Radical Mechanism
Analysis of the System and the Reactions Involved
In a free radical copolymerization, the number of chain radicals to be considered is given by the number of monomers present and the reactivity of a chain radical is practically determined by the free radical bearing terminal monomer unit and the size and chemical nature of those preceding the radical bearing terminal unit in the chain is considered to be of little consequence in this context.
In a binary copolymerization, therefore, two different chain radicals can be identified and each radical being capable of adding both the monomers, though not usually or necessarily with equal or comparable ease, the system is characterized by four simultaneously occurring propagation reactions. With two different kinds of chain radicals, and considering termination taking place only bimolecularly, the process is associated with three different chain termination reactions along with the possibility of several chain transfer reactions. Chain initiation takes place by reaction of primary radicals (derived from decomposition of the radical initiator used) with either of the monomers present with almost equal ease and efficiency in general or else, in the absence of an initiator, by thermal or photo activation of either monomer with different relative ease depending on the chemical nature of the monomers used.
Kinetics of Chain Propagation in Binary Copolymerization and Copolymer Composition
The four chain-propagation reactions in a binary copolymerization may be described by the reaction scheme (5) shown below:
k1,1 + M1 ® M1
k1,2 + M2 ® M2
k2,2 + M2 ® M2
k2,1 + M1 ® M1
Here, M1 and M2 are the two monomers, and M1 and M2 represent the chain radicals of all sizes with free radical bearing terminal units M1 and M2, respectively. k1,1 and k1,2 are rate constants for homopropagation and cross-propagation reactions respectively involving M1; likewise, k2,2 and k2,1 refer to the homopropagation and cross-propagation rate constants respectively involving M2. For a given rate constant, the first numeral is indicative of the reacting chain radical and the second numeral indicates the reacting monomer. Homopropagation relates to reaction between like radical and monomer and cross propagation relates to reaction between unlike radical and monomer.
Neglecting monomer consumption in the initiation step, the rates of consumption of the two monomers are written as
-(d[M1]/dt)=K1,1[M1][M1]+K2,1[M2][M1]
-(d[M2]/dt=K1,2[M1][M2]+K2,1[M2][M2]
An expression describing relative incorporation of the two monomeric units in the copolymer may be conveniently obtained by elimination of the radical concentration terms using the steady-state concept for each chain radical and then combining Eqs (6) and (7). Under steady state, the concentration of M1 and M2 remaining constant, the rates of interconversion of chain radicals must be equal, which prescribes that rates of reactions (ii) and (iv) of scheme (5) are equal, i.e.,
Equation (13) is known as the copolymer composition equation or simply the copolymer equation relating the copolymer composition and feed monomer composition with the help of the kinetic parameters r1 and r2, commonly called the monomer reactivity ratios. Equation (l3) clearly expresses that the copolymer composition is dependent on the molar ratio of the two monomers in the feed (but not actually on the absolute concentrations of the two monomers) and on the kinetic parameters r1 and r2, i.e., the monomer reactivity ratios. The copolymer equation may also be expressed as:

Significance of Monomer Reactivity Ratios
Examination of one or the other forms of copolymer equation clearly shows that with a knowledge of the feed monomer composition, the copolymer composition may be easily calculated if the monomer reactivity ratios r1 and r2 are known. The value of r1 ] 1 means that the chain radical has a preference to add the like monomer, i.e., M1 over the unlike monomer, M2, and r1 value [ 1 means just the opposite. The same concept applies to r2, which gives relative preference of chain radical M2 to add the like monomer (M2) over the unlike monomer (M1).[/P]
The absence of the initiation and termination rate constants in the copolymer composition equation, makes it readily apparent that the (instantaneous) copolymer composition is independent of rate of initiation and hence of the overall rate of polymerization and of initiator concentration. The composition is also not measurably affected in most cases due to the presence of solvents or other additives, indicating that the kinetic factors controlling the copolymer composition, i.e., the monomer reactivity ratios r1 and r2, remain unaffected by variations in initiator concentration, the overall rate of reaction and by the presence of various additives and solvents. They also usually remain unaffected even in heterogeneous systems as long as the reacting monomers are evenly distributed between the phases. However, the composition of copolymer would vary for differential distribution of monomers between phases. The values of r1 and r2 also change significantly for a drastic change in the mechanism of chain propagation from free radical to ionic, and even for ionic mechanism, from cationic to anionic mode of chain propagation. The copolymer equation has been found applicable to almost all comonomer combinations and equally applicable and relevant to radical, cationic and anionic systems, despite wide variations in r2 and r2 values depending on the mode of chain propagation. In radical copolymerization, the effect of temperature on monomer reactivity ratios is small or marginal in view of relatively low activation energies of radical propagation reactions and small differences in activation energies of different propagation reactions.
3. Types of Copolymerization
Ideal Copolymerization
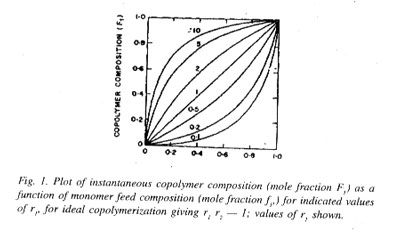
It is the widespread observation and experience that the mole fraction F1 is seldom equal to f1 and as a consequence of this feature, both f1 and F1 keep on changing with progress of copolymerization. The copolymer produced over a specified time period or range of conversion will consist of a mixture of copolymer molecules differing progressively in their compositions (F1).

In Fig. 1, plots of F1 vs fi for several cases of ideal copolymerization are shown and the values of r1 (= 1/r2) for each curve are given. For r1 = 1, the composition curve reduces to a straight line given by the diagonal and representing a special case in which r1 = r2 = 1 and hence F1 = f1 For r1 =r2 = k1.1 = k1,2 and k2,2 = k2,1 or in other words, the two monomers are equalry reactive with each radical and the copolymer composition, arising out of a random placement of the two monomers along the chain molecule, is equal to the feed monemer composition over the full range of monomer composition. In case of r1 or r2 i.e., for r1 ] 1 and r1 [ 1, or r1 [ 1 and r2 ] 1, one of the reacting monomers is more reactive than the other toward the two propagating species, and consequently, the copolymer formed will incorporate randomly a higher proportion of the more reactive monomer in its chain structure. The copolymer is proportionately richer in M1 in comparison with the feed monomer mixture for r1 ] 1, and just the opposite composition characteristics would result for r2 ] 1, i.e., for r1 [ 1. With large differences in the reactivities of the two monomers, (i.e., in the r1 and r2 values) the formation of copolymer sequences becomes progressively difficult as a consequence of very fast depletion of the more reactive monomer from the monomer mixture. Thus, for r1 = 0.1 and r2 = 10, giving r1r2 = 1.0 copolymers incorporating good amounts of M1 in its structure is difficult to obtain.[/P] 

shown in chain sequence (20) irrespective of the feed monomer composition. Copolymerization of stilbene (C6H5-CH=CH-C6H5) or of allyl acetate with maleic anhydride may be cited as examples closely resembling the case of alternation in copolymerization. Almost a perfect brand of alternation is shown by copolymers of sulphur dioxide and certain olefins, cycloolefins or vinyl compounds; they are commonly known as polysulphones and are obtained by radical polymerization of the respective monomer in liquid sulphur dioxide. The alternation is thought to arise as a consequence of self-polymerization of the 1 : 1 complex formed in situ between the olefin or related monomer (donor) and sulphur dioxide (acceptor).
Azeotropic Copolymerization
Some theoretical curves showing copolymer compositions (F1) for various feed monomer compositions (fi) corresponding to different r1 and r2. values, where r1r2 [ 1, (and also for some hypothetical cases given by r1r2 ] 1) are shown in Fig. 2. It may be seen that for both r1 and r2 [ 1 (or ] 1), the curves intersect the diagonal, i.e., the line representing F1 - f1. The point of intersection for a given system corresponds to a condition in which polymerization proceeds without change in composition and the composition of the polymer being formed is just the same as that of the monomer mixture at that point. Copolymerization of these critical mixtures of monomers is commonly known as azeotropic copolymerization. The critical monomer composition is then given by:
4. Average Copolymer Composition
The average composition of a binary copolymer obtained by polymerization of the two monomers M1 and M2 together over a finite range of conversion is conveniently calculated following the method and approach of Skeist. If [M] is the total or overall moles of the two monomers in the initial mixture, then [M] = [M1] + [M2], and initially [M1] is f1 [M]. For d[M] moles of monomers converted to polymer, the number of moles of M1 polymerized is F1d [M]. As during the process f1 changes by df1, and considering F1 ]f1, i.e., the copolymer formed is richer in M1 than the feed monomer mixture, the unreacted monomer mixture will contain (f1 - df1) ([M] - d[M]) moles of monomer M1. The moles of M1 going into the copolymer structure being equal to the difference in the moles of M1 in the monomer mixture before and after the reaction, one readily obtains
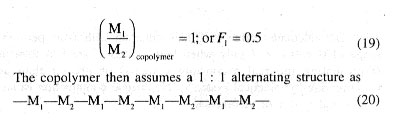
calculation of F1 as a function of f1 may be readily done with the help of Eq. (15) for a given set of values of r1 and r2. A knowledge of (F1 - f1) then permits graphical integration of Eq (4.25) to give the required degree of conversion {([M]o - [M])/[M]õ} where [M]õ is the initial value of [M], for a change in the feed composition from (f1)õ to f1. By successive application of this method for different values of f1 a relationship between f1 and degree of conversion may be obtained. The integral composition of the cumulative mass of copolymer formed during a given conversion stage is readily calculated from the shift in the value of f1 over the corresponding time span. This allows graphical expression of the instantaneous composition of the copolymer as a function of conversion.
Figure 3 shows how the average composition of the copolymer changes with degree of conversion for the styrene (M1) methyl methacrylate (M2) system for a case specified by (f1)õ = 0.80 and (f2)õ r1= 0.20 and r2 = 0.53 and n = 0.46. The instantaneous value of F is seen to follow f to unity (for M1) or to zero (for M2), but the average copolymer composition is seen to change marginally in comparison, becoming slightly but progressively richer in styrene with conversion.
For laboratory experiments, copolymerizations done to low conversions (within 5%) are better understood with the help of Eq. (15), in view f and F values as well as the average copolymer composition remaming practically unchanged over the limiting conversion zone. But this is impractical for an industrial process where replacement or replenishment of the depleting monomer at regular intervals is usually practised to minimize or eliminate variations in the monomer composition. For styrene-methyl methacrylate system, the reactivity ratios are quite close; so, it gives rise to rather limited variations in the average copolymer composition. But even such systems are also subject to the question as to just how much variation in the average composition can be tolerated without detrimental effects on the useful or desirable properties of the copolymer product. The replenishment of the depleting monomer (i.e., the more reactive monomer) often through continuous addition is usually worth the added cost that it entails.
5. Determination of Monomer Reactivity Ratios
For experimental evaluation of the monomer reactivity ratios, r1 and r2 copolymerization of the two monomers M1 and M2 at a number of feed compositions are done to low conversions, usually within 5%; the copolymers formed are then isolated, purified usually by repeated precipitation and finally analyzed for their compositions employing elemental analysis, determination of reactive groups, tracer (radioactive) technique, spectroscopic methods, etc. The feed and copolymer composition data may then be treated according to the following approaches for evaluation of r1 and r2:
(i) Trial and error or curve-fitting method. The method utilizes the experimental data on theoretical curves (F1 vs f1) to find which theoretical curve gives the best fit on trial selection of r1 and r2 values. This turns out to be a poor and tedious approach and precise determination of r1 and r2 is often difficult in view of insensitiveness of the composition curve to small changes of r1 and r2.
(ii) Method of intersection. For this method Eq. (13) is rearranged to express r2 in terms of r1 by the linear relationship given by Eq. (26):
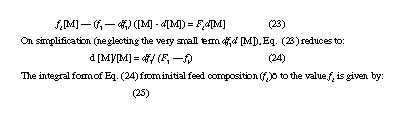
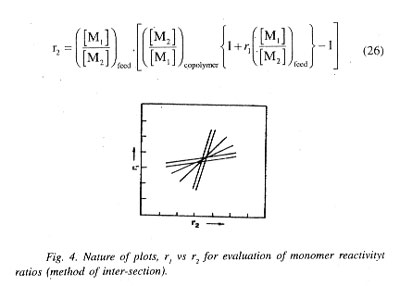
Plot of calculated values of r1 for assumed values of r2 or vice-versa for each experiment with a given feed composition gives a straight line. Theoretically, different linear plots for different experiments would intersect at a common point to allow evaluation of the r1 and r2 values for the given monomer pair from the intersection point, Fig. 4. In case there are experimental errors, the lines may not intersect at a single point and in that event the actual r1 and r2 values are obtained by averaging or from the central point of the contour given by the different points of intersection.
With known values of f1 and corresponding F1 values determined by experimental analysis, left hand side of Eq. (27) when plotted against the coefficient of r1 would yield a linear plot and the r1 and r2 values for the monomer pair would be given by the slope and intercept of the plot, respectively.
(iv) The method of limiting feed composition. Success of this method depends on finding very sensitive method of analysis of composition of copolymer prepared under conditions of very low values of ([M1]/[M2])feed such that the feed ratio progressively approaches zero. Equation (4.13) or its alternative form, Eq. (26), would then reduce to a more simple form given by Eq. (28). Likewise, the ratio given by feed composition ([M2]/[M1])feed
to copolymer composition ([M2]/[M1])copolymer may be equated to r1 for [M2] 0. Plot of the left hand side of Eq. (28) aganist the feed ratio, ([M1]/ [M2]feed yields a straight line passing through the origin and r2 is calculated from the slope of the linear plot. Calculation of r1 then readily follows from a knowledge of r2.
Monomer reactivity ratio of some selected binary copolymerization systems are listed in Table 1.
Rate of Copolymerization
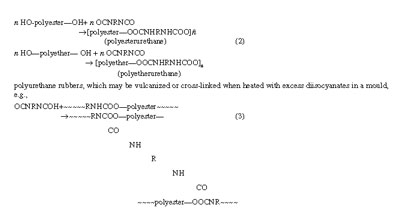
For treatment of copolymer composition, it was sufficient to consider the four propagation reactions or the two relative propagation rate constants, i.e., the monomer reactivity ratios, r1 and r2 for a binary system. Evaluation of overall rate of copolymerization requires consideration of the rates of initiation (R1) and termination (Rt) at the same time. Primary radicals released into the monomer mixture through initiator decomposition react with each monomer with nearly the same ease and efficiency so that consideration of two separate types of initiation is not necessary. Three types of termination must be considered assuming bimolecular mechanism involving all possible pairs of radical types. Under steady state condition of all the chain radicals, Ri being equal to Rt, one may write:
The values simply represent the reciprocals of the well known kinetic parameter kp /(2kt)1/2 for homopolymerization of the individual monomers and represents the ratio of half the cross-termination rate constant to the geometric mean of the termination rate constants for like radicals; ] 1 indicates that cross-termination is favoured and [ 1 means just the opposite. The factor 1/2 appearing in the ratio indicates that termination involving pairs of unlike radicals is statistically favoured by a factor of two compared with termination involving pairs of like radicals.[/P]
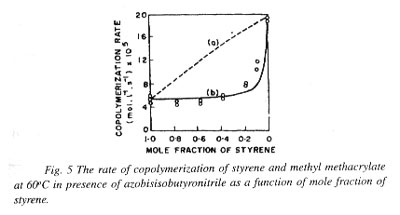
The values of d1 and d2 can be conveniently obtained from the studies of rates of homopolymerization of M1 and M2 using the general relation (-d ln[M]/dt = R1,2/d). The monomer reactivity ratios r1 and r2 are readily determined from studies of copolymer composition. A measurement of rate of copolymenzauon, then, allows calculation of f using Eq. (30a). A falling trend in the rate of copolymerization as compared to the mean of the rates of homopolymerization of the two monomers used follows as a consequence of the usual preference for cross-termination in copolymerization. Figure 5 shows how rate of copolymerization varied with feed monomer composition for a binary monomer system involving styrene and methyl methacrylate for (a) f = 1, showing the theoretical curve and (b) f = 13, showing the experimental points and the theoretical curve. The widespread experience is that the experimental rates of copolymerization fall below the f = 1 line, indicating that f is usually ] 1 and that the cross-termination rate exceeds the geometric mean for rate of termination involving like radicals.

7. Structure and Reactivity of Monomers and Radicals
Nature and number of substituents and the site of the substituents in case the unsaturated (olefinic) monomer bears more than one substituent on the unsaturation have decisive roles to play in determining the reactivity or stability of a specific monomer and the corresponding radical. The substituents influence or modify reactivity or stability by three basic approaches: (i) by activating or deactivating the p bond and thus rendering the monomer less stable and hence more reactive or vice versa, (ii) by imparting enhanced or reduced stability to the derived radical depending on better or limited prospects for resonance, and (iii) by providing different degrees of steric hindrance at the reaction site.
Structure and Reactivity of Monomers
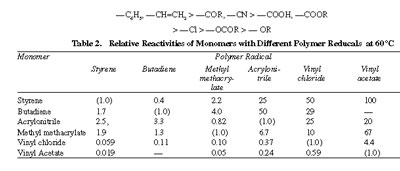
A relative order of reactivities of a series of monomers (Mz) is conveniently obtained by comparing the reciprocal monomer reactivity ratio (1/r1 = k1,2/k1,1) for each of the monomers in the given series with a specific chain radical,. If the rate constant for the reaction of the reference radical with its own monomer is taken as unity, relative monomer reactivities can be readily examined and compared. Related data for a few monomer-radical combinations are listed in Table 2. Thus, the first column under polymer radical of Table 2 compares reactivities of different monomers toward a chain radical having styrene unit as the radical bearing terminal unit, the second column gives a comparative idea of the reactivities of different monomers toward butadiene chain radical and so on. In view of different basis taken for each radical, values for different radicals given in different columns cannot be compared. The order of monomer reactivities is roughly parallel for all radicals, with some exceptions, here and there. The general order of enhancement of reactivity of a monomer by a substi-tuent toward radical attack is as follows:

Resonance Stabilization
The relative order of monomer reactivities toward a radical generally corresponds to the relative resonance stabilization of the radical formed on addition of the monomer. The more reactive monomer generally yields a radical of higher stability through resonance. A second substituent on the same carbon atom usually leads to an additive effect on the monomer reactivity or on the resonance stabilization of the corresponding radical.
Comparison of reactivities of different radicals toward the same monomer is conveniently done by making use of the absolute propagation rate constant, k1,1 and the reactivity ratio r1. Division of k1.1 by r1 gives an indirect measure of the rate constant k1,2 for reaction between radical and monomer M2. Absolute (propagation) rate constants for several redicals with each of the respective monomers are listed in Table 3. It is clearly seen that the reactivity of the radicals is in the reverse order of that for the monomers. Among the vinyl monomers shown, styrene is the most and vinyl acetate is the least reactive, while styrene radical is the least and vinyl acetate radical the most reactive. The styrene radical owes a high order of stability due to the possible existence of three quinonoid resonance structures, and as
The substituents also tend to stabilize the monomer, but the degree ot stabilization is much smaller for the monomer than for the radical. The resonance stabilization due to conjugation amounts to only about 3 kcal/ mol for styrene and butadiene as against a resonance stabilization to the tune of about 20 kcal/mol for the corresponding radicals.
Radical Reactivity and Steric Effects
The reactivities of different radicals toward a given monomer and vice-versa can be judged and appreciated from a comparison of the respective reaction rate constants listed in Table 3. Quite expectedly, a substituent rendering the monomer more stable confers on the corresponding radical higher reactivity and vice-versa. A substituent's role in enhancing the reactivity of a radical is much greater than its effective role in depressing the reactivity of the monomer. Thus, the vinyl acetate radical is apparently 500-1500 times more reactive than the styrene radical toward a given monomer, but the styrene monomer is only about 50-100 times more reactive than the vinyl acetate monomer. If and represent an unstabilized and a stabilized radical and M and Ms represent an unstabilized and a resonance-stabilized monomer, then the radical-monomer reactivity follows the order:
(R + Ms) ] (R + M) ] (Rs + Ms) ] (Rs + M)
Substituents are also known to influence the radical monomer reaction rate constants from the view point of steric hindrance. This can be well appreciated by considering some of the related rate constant data for monomers such as vinyl chloride (monochloroethylene), vinylidene chloride (1,1-dichloroethylene), 1,2-dichloroethylene and polychloroethylenes, given in Table 4. The effect of a second substituent on the monomer reactivity produces roughly an additive or reinforcing effect for the 1,1-disubstituted monomer, but in the case of a 1, 2 disubstituted monomer, there is a significant decrease in reactivity principally due to steric hindrance.
Table 4. Rate Constants k1,2 for some Radical-Monomer Reaction at 60oC Showing Steric Effect of Substituents in Copolymerization (1 mol -1g-1)
<th colspan='2' align='center'>Radical (M1)
| Monomer (Mg) |
| |
Vinyl Acetate |
Styrene |
| Vinyl chloride |
43,000 |
10.35 |
| Vinylidene chloride |
1,00,000 (68°C) |
95.14 |
| cis-1,2-Dichloroethylene |
1,580 |
0.83 |
| trans-1,2-Dichloroethylene |
9,900 |
4.75 |
| Trichloroethylene |
15,200 |
11.0 |
| Tetrachloroethylene |
1,470 |
0.95 |
k1,2 (= k1,1/r1) values were culculated using k,1,1, i.e, kp value for vinly acetate as about 10,000 and for styrene as 176 and the respective r1 values from the literature.
Among the two 1,2-dichloroethylenes, the cis-isomer is much less reactive than the trans-isomer toward reaction with a given radical in copolymerization because the cis-isomer is less stable and also because the cis-isomer is unable to achieve a completely coplanar conformation in the transition state during reaction with a radical, thus impairing resonance stabilization of the derived radical by the substituent.21
The lower reactivity of tetrachloroethylene in comparison with that of trichloroethylene is analogous to the difference in reactivities between vinyl chloride and 1,2-dichloroethylene. Trichloroethylene is more reactive than the 1,2-dichloroethylene but is less reactive than the 1,1-dichloro derivative i.e., vinylidene chloride.
Polar Effects and Alternation
Examination of Tables 2 and 3 reveals that the reactivities of a given monomer to different radicals or of a given radical to different monomers do not exactly follow the same order for different reference monomers or for different reference radicals respectively. Monomer and radical reactivities are interdependent. Deviation of the product r1.r2 from unity and its closeness to zero is taken as a measure of alternating tendency and this enables one to tabulate monomers in order of their r1r2 values with other monomers, as shown in Table 5 in a diagonal manner such that further apart the two monomers are the greater is their tendency to alternate. Monomers with electron releasing type of substituents are placed at the top and those with electron withdrawing substituents at the bottom part of the table. Thus, a specific nature of radical-monomer reaction manifested through alternation is favourably achieved for monomer pairs with dissimilar polarization properties of the substituents.
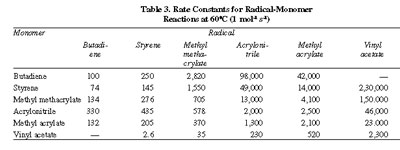
Thus, acrylonitrile copolymerizes randomly (ideal copoly-merization) with methyl vinyl ketone (r1r2 = 1.1) while it undergoes alternating copolymerization with alkyl vinyl ethers (r1r2 = 0.0004). Monomers which do not homopolymerize will readily copolymerize with strict regularity of alternation, as exemplified by the copolymerization of stilbene (electron donor) and maleic anhydride (electron acceptor). Such copolymerization stands as a fine illustration of polar effect and alternation.
Alternating or 1 : 1 copolymer may also result more as a consequence of 1 : 1 donor-acceptor complex formation between electron-donor and electron-acceptor monomers and homopolymerization of the complex by a head to tail mechanism than due to the alternate addition of the donor and acceptor monomers to the propagating radical. Catalyzed or spontaneous polymerization of suitable olefins or cycloolefins (donor) in liquid sulphur dioxide (acceptor) to produce 1 : 1 olefin/cycloolefin-sulphur dioxide copolymers is believed to proceed via initial 1 : 1 donor-acceptor complexation and homopolymerization of the complex in the subsequent step.

8. The Q - e Scheme
In an attempt to correlate structure and reactivity of radical-monomer systems, various approaches have been developed and tried, including the Hammett a- approach, with fair decree of success. The Q-e scheme of Alfrey and Price developed for this correlation is of more general applicability. According to this scheme, the rate constant k1,2 for copolymerization involving reaction between radical and monomer is written as
k1,2 = P1 Q2 exp (-e1e2) (32)
where P1 and Q2 relate to the general reactivities and e1 and e2 give measures of the polarities of the radical and monomer M2 respectively. Resonance effects in the radical and the monomer are defined and expressed by the P and Q terms and assuming that the same e value applies to both monomers, M and its radical, M one can write
k1,1= P1Q1 exp ( - e2/1) (33)
k2,2= P2Q2 exp ( - e2/2) (34)
k2,1= P2Q1 exp ( - e2e1) (35)
r1= (Q1/Q2 exp [ - e1(e1 -e2)] (36)
r2 = (Q2/Q1 exp [ - e2(e2 -e2)] (37)
r1/2 = exp [-e1 -e2)2] (38)
Thus, the Q-e scheme correlates monomer-radical reactivities using the parameters Q1, Q2, e1 and e2. Reactivity ratios for copolymerization of any pair of monomers can be computed by assigning appropriate Q and e values to each set of monomers. However, because of its semiempirical approach and the associated uncertainties in the assignments of Q and e values, the Q-e scheme is subject to much criticism on theoretical grounds. The assumption that the same e value applies to both the monomer and the radical derived from it is made without much justification. Moreover, the concept that alternation in copolymerization is electrostatic in origin is not largely supported by experiments, as the alternating effect does not generally depend on the reaction medium. Thus, the electrostatic basis or polarity concept behind the Q-e scheme and the equations based on the scheme should be judged and used with all these limitations in veiw. Despite all criticisms, the Q-e scheme has proved quite useful as a basis for correlating a wide expanse of otherwise incoherent kinetic data (r1, r2, k1,2 and r1r2) relating to radical monomer reactions in a satisfactory and even splendid manner. The Q-e scheme may be considered to be an effective transcription to equation form of the reactivity and polarity series of Tables 2 and 5. The scheme has also been conveniently employed for correlation of data relating to chain transfer involving a transfer agent and a radical.
In conclusion it may be said that irrespective of the approach or basis of analysis and interpretation adopted, the reactivity of a monomer in copolymerization depends primarily on two factors: (i) The first factor relates to the intrinsic behaviour of the monomer (resonance factor). The order of reactivity of the monomer is determined most significantly by its capacity for resonance stabilization in the transition state, (ii) The second factor is, however, connected with the specific interaction between a monomer-radical pair (polar effect). The second factor modifies the first in a manner that depends roughly on the degree of dissimilarity in the polarization of the two reacting species --the higher the dissimilarity, the more favourable the reaction.
9. Technical Significance of Copolymerization
The modification of a polymer by introducing a second monomer during the polymerization process has long been used to alter its physical properties and sometimes to achieve improved processing characteristics. Incorporation of a second monomer structure as repeating units in the polymer, i.e., the copolymer molecules, reduces overall molecular symmetry leading to lowering (or loss) of degree of crystallinity and of softening temperature. Copolymerization is therefore, considered as a case of internal plasticization. The modification of poly (vinyl chloride), (PVC) by taking small proportions of vinyl acetate (VA) monomer with vinyl chloride (VC) during polymerization to make more flexible and soluble resins of easier processability is well known. Similarly, straight polybutadiene has many technical disadvantages arising from its poor tack and difficult processability. But Copolymerization of butadiene with styrene, acrylonitrile and other monomers has led to many technically important resinous and rubbery products. There is practically unlimited scope for variation in polymer structure and polymer properties through copolymerization. In fact, the number of technically important copolymers far surpasses that of important homopolymers. Besides the synthesis of copolymers having random or alternate sequences of monomeric units which have so far been considered and discussed, it is also possible to recognize and synthesize copolymers having two other different kinds of sequences, and they are commonly known as block and graft copolymers.
Polymer Characteristics and Polymer Characterization
1. The Structure of Vinyl and Related Polymers
The selective nature of ionic polymerization in respect of the nature of monomer, solvents and additives, the complexities and variations in the mechanistics and kinetics depending on the nature and identity of the ionic catalyst including the different structural (steric) sequences imparted under the influence of different gegen ions are generally established and widely recognized. For all these and associated reasons, ionic polymerization is not favoured and widely adopted for industrial polymerization. It is employed only in a few instances where the objective is the synthesis of a polymer of high structural (steric) regularity. Most industrial polymerizations of vinyl and related monomers are done by free radical means because of many practical and economic advantages and versatility associated with the free radical process.
2. Prevalence of Head-to-Tail Structure in Vinyl Polymers
Addition of a free radical R. (primary radical or chain radical) to a vinyl monomer may take place according to either of the two ways as shown below:
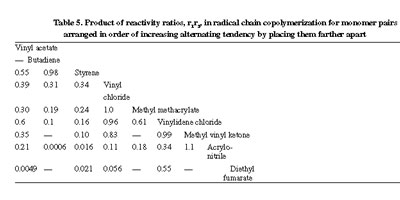
The relative stabilities of the product radicals I and II determine the relative rates of reactions (1) and (2). In structure I, the presence of the substituent X on the radical-bearing carbon atom ( -carbon atom of the monomer) offers a much higher scope for stability of the radical and the degree of stabilization would depend to a large extent on the capacity of the substituent for resonance. The scope for such stabilization is poor in structure II in view of the non-availability of the substituent for taking part in resonating structures involving the lone electron as the substituent is attached to the carbon atom in the -position with respect to the radical site. Moreover, on consideration of steric hindrance to the approaching radical offered by the substituent X of the vinyl monomer, reaction (1) appears to be more consequential than reaction (2). The steric factor would, however, be dependent on the size of the substituent. Successive addition of monomers exclusively by the favoured reaction route (1) in the propagation step would result in a head-to-tail structure, i.e., the polymer formed would exhibit 1, 3 structure with respect to the placement of the substituent, X, along the chain axis. For monomer addition exclusively by reaction route (2), the resultant polymer would also exhibit head-to-tail or 1,3 structure (III):
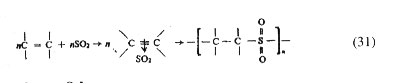
On the other hand in an extreme case where monomer addition is permitted alternately by routes (1) and (2), the resultant polymer would appear as having head-to-head, tail-to-tail or 1, 2-1, 4 structure (IV).

But from different theoretical considerations, the head-to-tail structure is expected to be the prevalent structure for all cases where the monomer structure is such as to permit addition of the chain radical to the monomers by only one route to the virtual exclusion of the other. For monomers with a substituent X producing little directive influence, random addition giving all kinds of possible arrangements of the repeat units in the polymer chain (1, 3 and 1, 2-1, 4 arrangements) may take place.
However, the structural pattern of vinyl polymers, as confirmed by different experiments using polymers of different monomeric origin, appears to be mostly head-to-tail in nature. Pyrolysis or destructive distillation of polystyrene at about 300°C produced 1, 3-diphenylpropane (V), 1, 3, 5-triphenylpentane (VI) and 1, 3, 5-triphenyl-benzene (VII).
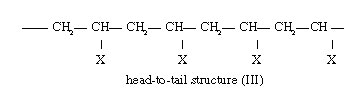
In these products the phenyl groups are attached to alternate carbon atoms as in polystyrene having head-to-tail structure and no products having phenyl groups on adjacent carbon atoms are obtained.
Treatment of a dilute solution of poly(vinyl chloride) in dioxane with zinc dust leads to removal of chlorine producing cyclopropane rings along the chain. However, a maximum of about 84-86% of chlorine can be normally removed in this manner. The retention of nearly 14% chlorine on extended reaction is attributed to the occasional isolation of a lone chlorine substi-tuent between neighbours that have paired at random and reacted:
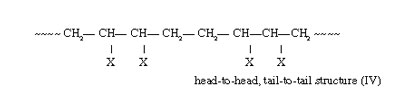
The observed extent of haiogen removal is in good agreement with the value (86.5%) obtained by statistical calculations assuming head-to-tail structure. Successive head-to-head, tail-to-tail structure (IV) without a break in the sequence, on the other hand, would have produced a polyene structure (-CH2-CH = CH-CH2-CH2-CH = CH-CH2-); for a random structure, however, theoretical calculation predicts that 18.4% of chlorine should escape elimination and would be retained in the polymer, considering that chlorine atoms in 1, 2 and 1, 3 positions only are removable in pairs by zinc
Likewise, poly(methyl vinyl ketone) transforms into a product consisting, of long sequences of condensed cyclohexene rings interrupted by statistically isolated groups at random intervals through internal aldol condensation, much in conformity with a head-to-tail structure for the polymer. Extended reaction results in progressive removal of oxygen for which the experimental upper limit is about 79-85% against a calculated value of 81.6% for intramolecular reaction of the kind in a head-to-tail polymer. For 1, 2 placements of all successive pairs of the substituents leading to formationof 1, 2 and 1, 4 diketone structures
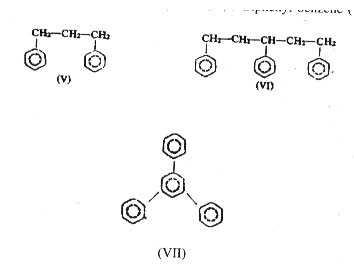
In a similar manner, poly (vinyl acetate) also appears to be largely characterized by a head-to-tail arrangement of the repeating units in the chain molecules as revealed by treatment of the hydrolyzed polymer, i.e., poly(vinyl alcohol) with periodic acid reagent which attacks only the 1, 2-glycols and gets consumed in the process. Experimental reports, however, indicate little consumption of the reagent substantiating successive monomer addition by head-to-tail mechanism despite the fact that the substituent-OCOCH3 offers very limited stability to the free radical through resonance. At the same time, periodic acid or periodate ion treatment in aqueous solution causes rather notable lowering in molecular weight of the poly(vinyl alcohol) within a few minutes at room temperature, giving evidence for a minor proportion of head-to-head/tail-to-tail structure. It is important to bear in mind that even for head-to-tail propagation sequences, termination by combination results in a break in the monotony of the 1, 3 structure and causes appearance of a 1, 2 structure at the point of radical combination.
3. Branching in Vinyl Polymers
Consideration of an olefinic unsaturation as equivalent to bifunctionality leads one to ordinarily presume the vinyl and related polymerizations as processes yielding linear macromolecules. But in radical polymerization both the monomers and the polymers being formed or present in the system are subject to attack by propagating radicals through what may be termed as monomer transfer and polymer transfer reactions, thereby leading to formation of some branched polymer molecules in the product. In certain cases, such as in vinyl acetate, chain transfer with the monomer may lead to a polymer molecule with a terminal unsaturation as shown in Eq. (6):
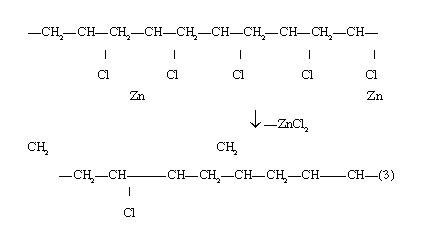
Polymer transfer during polymerization of a given monomer may sometimes lead to different degrees of branching; and in selected systems extensive chain transfer with the monomer and with the polymer formed may lead to extensive branching and even to limited cross-linking, particularly when the polymerization is allowed to proceed to high conversions. Among the common monomers widely studied, extensive long-chain branching is the characteristic feature of vinyl acetate polymerization. Polymerization of ethylene by the high pressure (free radical) process leads to extensive short chain branching as a consequence of intramolecular chain transfer.
The polymerization of conjugated diolefins and related diene monomers tends to yield partly insoluble or even largely insoluble polymer products due to limited or extensive cross-linking involving the residual or rearranged unsaturation in the repeat units. It is quite obvious that the proportion of insoluble polymer or the gel content increases with degree of conversion. Soluble and industrially useful polymers are usually prepared by limiting the degree of conversion and often by controlling the chain length using calculated molar proportions of a chain transfer agent commonly known as a regulator.
One sure means of producing cross-linked polymers is the use of small or large proportions of a divinyl compound as a comonomer with a (mono)-vinyl monomer, typically exemplified by the copolymerization of styrene with a small amount of divinyl benzene. Examples of other bi- or poly-functional (olefinic) monomers used as cross-linking agents are ethylene glycol dimethacrylate, triethylene glycol dimethacrylate, glyceryl trimethacrylate, triallyl cyanurate, diallyl sebacate, etc.
4. Polymer Degradation
By the term degradation, we generally mean breaking down of chemical structure of a given material to smaller fragments, and with respect to polymeric materials it simply implies a decrease in molecular weight or chain length. But sometimes, we may even see a polymer lose its useful or desirable properties and in the process it gains in molecular weight. It is difficult to give a close definition of polymer degradation. In a broad perspective, any deterioration of properties of polymers which make them variously useful as rubbers, plastics or fibres should be considered as a case of degradation. Discolouration or embrittlement of a polymer is a degradation process even though it may not lead to chain breakage or any fall in molecular weight. A degradation process may result in no measurable change or different degrees of decrease or increase in molecular weight, depending on the nature of the polymer and its environments.
In most degradation studies, however, the focus of attention is on: (a) change in molecular weight, (b) change in physical and mechanical properties, (c) evolution of volatiles and their chemical identity, and (d) the chemical structure of the residual polymer vis-a-vis the same of the original polymer. Almost in all practical applications it is our objective and desire that the polymer performs up to expectations and in tune with its potentials without degradation due to the various agencies inherent or given rise to in its environments, such as, heat, light and other radiation, atmospheric oxygen, moisture, acid, alkali, and other chemicals, mechanical forces, micro organisms, etc. Even if a polymer material degrades, the degradation process can be possibly prevented or delayed and the polymer made more resistant to the degrading agent with the use of certain additives only if one has a precise understanding of the mechanism of the degradation reactions. As for example, when the role of oxygen was recognized in the deterioration of polymers, the relevance and importance of antioxidant additives were better realized and appreciated.
The backbone structures of different polymers tend to degrade by two distinct mechanisms by thermal or hydrolytic means, i.e., by a chain mechanism or by a step mechanism. Condensation or step-growth polymers are generally prone to hydrolysis (or thermal splitting) of the condensed inter unit linkages, such as the ester and amide linkages and the like, by a step mechanism (much alike the mechanism of their formation) to yield low molecular weight degraded products. Addition or chain-growth polymers, on the other hand, are usually prone to degradation under heat, light, high energy radiations or otherwise by a chain-mechanism to yield low molecular weight products.
The hydrolytic degradation of condensation polymers takes place randomly and it follows a pattern similar to that known for related low molecular weight compounds and hence, such degradations of specific condensation polymers are not taken up for discussion here. Drop in molecular weight is quite severe and drastic even for limited hydrolytic degradation of condensation polymers.
Thermal Degradation
Degradation of polymers at elevated temperatures has been most widely studied. The degradation mechanism for different polymers does not follow a uniform pattern. The thermal stability and mode of decomposition of a polymer are largely dependent on the chemical structure of the chain segments or repeat units. It is interesting to note that the degradation characteristics of a polymer cannot be precisely predicted on the basis of those exhibited by low molecular weight model compounds. Even though the mode of decomposition is much the same, the polymers degrade or decompose at temperatures far lower than those of the corresponding model compounds. The degradation may primarily lead to chain scission or involve non-chain scission reactions to begin with. The chain scission or depolymerization reactions result in cut down in chain length or molecular weight due to breaking of main polymer chain backbone. For a degradation of this nature, the products at any intermediate stage of the reaction are very much similar to the parent polymer in the sense that the characteristic monomer units can still be distinctly recognized. The non-chain scission reactions are basically substituent reactions in that the substituents attached to the main polymer chain backbone are partially or totally modified or eliminated. This leads to major changes in the chemical nature of the repeat units in the macromolecular structure and the volatiles liberated in such cases are chemically unlike the monomer.
Depolymerization
Depolymerization brought about thermally may be manifested as depropagation with elimination of monomers only as the volatile or by random breaking of the chain backbone giving rise to low molecular weight volatiles other than the monomer with or without the elimination of monomer in part Poly(methyl methacrylate) provides an example of one extreme system where depolymerization yields monomer almost quantitatively, and for such a system, decrease in molecular weight proceeds very slowly with the beginning of degradation and the fall in molecular weight is measurable only at some advanced stages of degradation or volatilization. Polyethylene on the other hand represents another extreme system that yields little monomer on depolymerization while resulting in sharp fall in the molecular weight to low values at quite small extents of volatilization.
The depolymerization mechanisms are far from simple and a number of reactions presumably take place simultaneously and consecutively. The initiation of chain degradation process may occur by random chain scission or it may be a case of chain-end initiation. The initiation reaction is followed by a depropagation reaction in selected systems, forming monomer. The overall depolymerization process may be schematically represented as follows:
1. Initiation of depolymerization
(a) Random chain scission
Pn Rn-i + Ri
(b) Chain-end initiation
Pn Rn-1+ R1
2. Depropagation
Rn Rn-1 + M
3. Chain transfer involving random chain scission
Rn + Pm Pn + Pm-i + Ri
4. Termination
Rn Pn unimolecular (obscure mechanism)
Rn + Rm Pn+m bimolecular (radical coupling)
Rn + Rm Pn+Pm bimolecular (radical disproportionation)
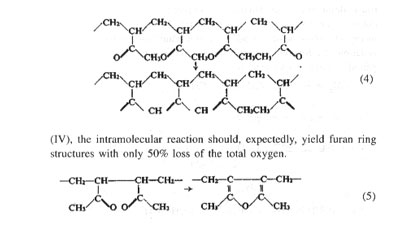
In scheme (8), P, R and M denote polymer, radical and monomer respectively and the subscripts, as usual, signify chain length or degree of polymerization. The monomer content in the volatiles obtained on depolymerization of several polymers are listed in Table l from which one can well appreciate how significantly the course of depolymerization is dependent on the structure of the repeat units of the polymer. Substituents modify the reaction course profoundly even if the main chain scission reactions prevail. For ethylene and 1-substituted ethylenes, non-chain scission reactions are predominant, producing either little or up to 50% monomer as volatiles. 1, 1-disubstituted repeat units apparently favour 100% monomer yield. Monomer yield is negligible for presence of chlorine substituent.
Substituent Roles
Substituents control the degradation process in many polymers containing labile a-hydrogen in the repeating units. Substituent reaction or substituent elimination often spares the main-chain C-C links from breakdown in such polymers. However, the substituent reactions have little comparable uniformity, each kind of substituent has its characteristic chemical nature and reactivity. The substituent reactions assume prominence if only they are initiated and accomplished at temperatures lower than that at which main chain bonds are broken. Evidently, then, substituent reactions are normally observed at comparatively low temperatures.
One of the more common polymers showing characteristic substituent reaction is the poly(vinyl chloride), PVC. Had PVC in reality corresponded to the idealized concept of regular repeating units it would have been expected to be a polymer of high inherent stability based on evidences available from studies of low molecular weight model compounds such as 1, 3, 5-trichlorohexane.12 Thermal dehy-drochlorination of PVC begins slowly at 80-100°C but it becomes quite rapid at ] 140°C. Evidences from many studies indicate that the dehydrochlorination of PVC is due initially to a loss of HC1 molecule which takes place at a site on the molecule either containing or adjacent to a tertiary or (terminal) allylic chlorine atom (arising in the polymer due to chain transfer by different mechanisms or termination by disproportionation), each of which potentially functions as an activating group or centre. Radical detection in the electron spin resonance spectra of dehydrochlorinated PVC is indicative of a radical mechanism for thermal degradation of PVC. However, it is unlikely that a radical chain process of any great propagation length is involved. It is more likely that a radical reaction of short chain length is involved as follows:
~~CH2-CHCl-CH2-CHCl-CH = CHCl
~~CH2-CHCl-CH2-CH-CH = CHCl + Cl
~~CH2-CHCl-CH = CH-CH = CHCl + HCl
The elimination of the first molecule of HCl and consequent formation of a double bond on PVC chain subsequently activates the neighbouring chlorine atom which assumes the nature of an allylic chlorine, causing elimination of another hydrogen chloride molecule, with the process continuing to repeat itself finally producing a conjugated polyene structure (VIII) by an autocatalytic effect.
~~CH2 = CH-CH = CH-CH = CH~~
Certain features of thermal dehydrochlorination of PVC, not compatible with a radical mechanism, can be explained and appreciated on the basis of ionic mechanism. Evidences in the studies of HCl "unzippering" from PVC lend support to the currently held theory that both radical and ionic processes may be at work simultaneously. The most effective and widely adopted means of arresting PVC degradation is through incorporation of chemical additives such as basic lead carbonate, tribasic lead sulphate, etc., which are basically HCl scavengers. They effectively react with HCl as and when liberated under processing or service conditions and inhibit the HC1 unzippering. Inverse proportionality of PVC decomposition rate to molecular weight stands in evidence for chain-end initiation.
Another good example of substituent reaction is found in the colouration of polyacrylonitrile or polymethacrylonitrile thermally at temperatures near and above 175°C. The colour reactions are mostly due to intramolecular linking of nitrile groups to form conjugated unsaturation of carbon nitrogen sequences (IX), forming ladder-type structure:
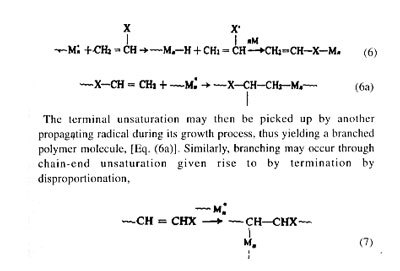
The reaction leading to colour formation is catalyzed by carboxylic acid, more particularly for polymethacrylonitrile. For polyacrylonitrile, the intramolecular reaction is occasionally intercepted by intermolecular reaction leading to insolubilization of the product even before development of visible colour. Polymethacrylonitrile, however, remains soluble even when extensively coloured, indicating exclusive or near-exclusive occurrence of intramolecular reaction for this polymer.
Mechanochemical Degradation
Breakdown of molecular chains under shear or mechanical force, often aided by a chemical reaction (oxidation) is called mechanochemical degradation. Mastication of rubber leading to chain breakage and development of plasticity under shear is a good example of mechanochemical degradation of polymers. It is a widespread experience that mastication of natural rubber at ordinary temperatures in an atmosphere of nitrogen brings about no appreciable change in molecular weight and development of plasticity. In air or oxygen, however, degradation is immediate and rapid. Under mechanical shear, the rubber molecules break into radicals, and oxygen, a radical scavenger, readily reacts with them leading to permanent chain breakage. In nitrogen, however, the primary radicals formed under shear immediately recombine to give no effective chain breakage. If benzoquinone, also a radical scavenger, is used in small concentrations (1 phr), rapid degradation even in nitrogen is observed, while little additional effect is exhibited in oxygen.
In natural rubber, the weakest bonds are those linking the different isoprene units as shown below, and scission of each such bond results in generation of two relatively unreactive radicals stabilized notably by allylic resonance.
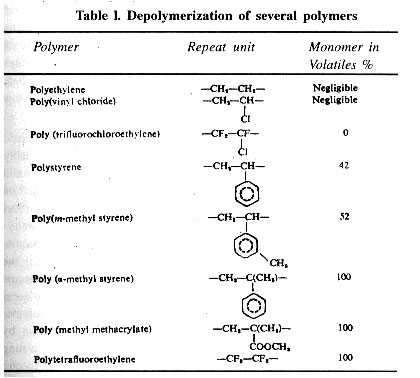
The radicals thus derived are so unreactive as to be practically unable to take part in disproportionation reaction required to lead to permanent chain breakage in absence of radical acceptors. For polymers having no backbone unsaturation, stabilization of primary radicals formed under shear in this manner are remote such that radicals formed due to bond scission under shear are very much more reactive and effective chain degradation takes place even in nitrogen.
Aging or Oxidative Degradation
Almost all polymers are subject to limited oxidative degradation, i.e., chain breakage subsequent to reaction with oxygen, particularly when processed at elevated temperatures, often under high pressure for short time intervals. Saturated hydrocarbon polymers, such as polyethylene, are very much more resistant to aging or oxidative degradation as compared to unsaturated hydrocarbon polymers, such as natural rubber or synthetic diene polymers under ambient conditions. Polymers bearing tertiary hydrogens in the repeat units or methylene groups in the a-position with respect to chain unsaturation are usually more prone to aging. The primary attack by oxygen leads to hydro-peroxidation and the hydroperoxides formed subsequently decompose radically under heat/light with the onset of a chain process that ultimately results in chain degradation.
For unsaturated polymers such as natural rubber, the mechanism of oxidation is more complicated and the process turns out to be an autooxidation process. Here, the hydroperoxidation by primary oxygen attack takes place in the a-methylenic position. Subsequent sequence of reactions involve (a) decomposition of the hydroperoxide into radicals and onset of an ill characterized chain degradation or cross-linking process, and (b) epoxidation involving double bonds and the primarily formed hydroperoxides. Subsequent decomposition of the reactive epoxides and further oxidation by oxygen cause chain breakage at olefinic bonds or even saturation of double bonds without chain rupture. The net result is incorporation of oxygenated polar groups such as -OH, -COOH, -CO- and -O- in the polymer. In any event, the loss of unsaturation does not follow oxygen intake quantitatively.
Photodegradation
Polymers are also subject to photodegradation to different extents depending on their chemical structures and certain contaminants that may be present in them. Degradation by light is rather selective in nature. Specific groups would absorb light of specific wave lengths and induce degradation. The power to photodegrade increases as the wavelength of the incident radiation decreases, i.e., as the energy of the quanta increases. Light of wavelength 2800 Ã... corresponds approximately to a bond energy of 100 kcal/mol, and the wavelength region of 3000-4000 Ã... corresponding to bond energy of nearly 90-70 kcal/mol is considered as the threshold. For light above this wavelength range, photodegradation scarcely occurs (a C-C single bond has energy in the region 80 kcal/mol and the C-H bond strength is in the region 80-100 kcal/mol depending on environment). Photodegradation is widely recognized to proceed via free radical mechanism. The bonds or groups which absorb energy and those which subsequently break into radicals and the course of radical chain degradation that follows largely depend on the chemical nature of the material and the energy of the incident radiation. High energy radiations such as X-rays, g-rays, b-rays, high speed electrons, etc., often bring about uncontrolled degradation usually involving simultaneous radical and ionic mechanisms.
5. The Concept of Average Molecular Weight
A given polymer material is mostly a mixture of molecules of (nearly) identical chemical structure but varying in chain length or molecular weight. The molecules produced in polymerization reactions have lengths that are distributed in accordance with a probability function which is governed by the mechanism of the reaction and by the conditions under which it has been carried out. The concept of average molecular weight is therefore important and relevant and the assignment of a numerical value to the molecular weight of polymer requires the definition of a particular average. An average molecular weight may be generally expressed as

Equation (15), in effect, can be expressed as a summation series resembling Eq. (13) where the fractional coefficients represent mole fractions of the respective molecular species present in the system, knowing that total weight W = S NiMi and total number of molecules N = S Ni. Thus,
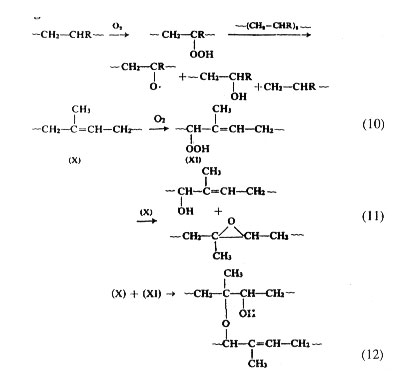
Here w1,w2, w3 etc., stand for weights of different species having molecular weights, M1, M2, M3, etc., respectively and S wt = W is the total weight of all the molecules present.
The consequence of definitions given above is that w n; the equality, however, corresponds to the limiting case of a perfectly monodisperse sample. The deviation from unity of the ratio w//n, known as the distribution ratio, is taken as a measure of polydispersity of polymer samples, a 'higher value of the ratio indicating greater polydispersity.
Evaluation of number average molecular weight is useful in understanding the polymerization mechanism and kinetics. The evaluation of n is also useful in the analysis of kinetic data to examine the effects of many side reactions including chain transfer, inhibition and retardation, and auto-acceleration or gel effect in vinyl polymerization. The number average molecular weight also assumes prime importance in determining the solution properties of the polymer, commonly known as the colligative properties. Polymer molecules of lower molecular weight contribute equally and enjoy equal status with those of higher molecular weight in determining these properties.
Weight average molecular weight, on the other hand, is important in relation to bulk properties of polymers that reflect their load bearing capacity. Softening, hot deformation, tensile and compressive strength, modulus and elongation, toughness and impact resistance and some other related bulk properties of polymer are better appreciated on the basis of weight average molecular weight, keeping in mind, however, the influence of chemical nature of the repeat units, degree of branching and cross-linking, thermal or thermomechanical history of the sample and other related factors in this context.
6. Viscosity Average Molecular Weight
The viscosity of a polymer solution (h) is higher than that of the pure solvent (h0) at a given temperature and the gain in medium viscosity on dissolving the polymer in the solvent is a function of both molecular weight and concentration of the polymer solute. Even though the solution viscosity is easy to measure, it does not give a direct and absolute value of molecular weight. If the polymer solution is very dilute and consequently the density change of the solvent due to dissolved polymer is negligible, then the viscosities of the solution and solvent at a given temperature would be proportional to their flow times in a capillary viscometer such that the relative viscosity, hr expressed as the ratio, h/hõ would be given by the flow time ratio t/tõ where t and tõ are the flow times of the solution and solvent respectively. Both relative viscosity (hr) and specific viscosity (hsp) defined as hsp= (h-hõ)/hõ are dimensionless. If the solute macromolecules do not interfere with one another during flow, the viscosity gain is proportional to their concentration and hsp/c, commonly known as the reduced viscosity, would be a constant. But for polymer solutions, hsp/c is generally found to increase with increase in c. The intrinsic viscosity or the limiting viscosity number, [h] for a given polymer-solvent syitem at a given temperature is given by the intercept of the plot of hsp/c vs c when the linear experimental plot is extrapolated to zero concentration, or more precisely, to infinite dilution condition. The concentration dependence of polymer solution viscosity is conveniently expressed by two empirical equations, known as Huggins' equation given by Eq. (19) and Kraemer's Equation given by Eq. (20), i.e.,

where K and a are constants for a particular polymer-solvent system at a given temperature. This equation provides a basis for determining molecular weight from viscosity measurements. The values of v obtained are not absolute in view of incomplete interpretations of K and a. The values of K and a must normally be determined by measuring the [h] values of monodisperse polymer samples whose molecular weights have been obtained from one of the absolute methods such as osmometry and light scattering and making use of a plot of log [h] vs log M. The exponent a varies with both the polymer and the solvent; its value usually ranges between 0.5 and 0.8. It does not fall below 0.5 in any case and exceeds 0.8 in exceptional cases particularly for polyelectrolytes in the absence of added salts.
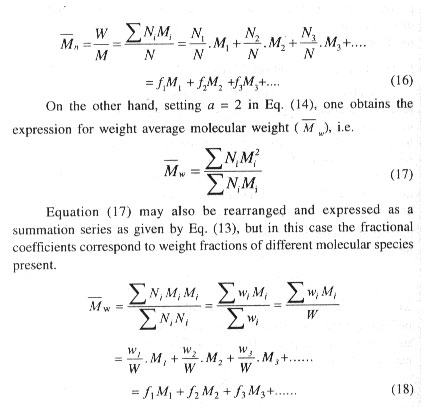
For most systems, K and a are best understood when [h] is determined in a solvent at what is known as the theta (q) temperature. The value of a is then equal to 0.5 and K depends on the temperature while remaining independent of the solvent, keeping in mind, however, that the solvent fixes the temperature of measurements. At the q temperature, the chemical potential due to polymer (segment)-solvent interactions is zero and the deviations from ideality just vanish. Therefore, the free energy of interactions of the segments within a volume element is also zero. In fact, q temperature is the lowest temperature for complete miscibility in the poor solvent used at the limit of infinite molecular weight. The ideality is struck at the q temperature because the molecular dimensions are unperturbed by intramolecular interactions.
General Expression for Viscosity Average Molecular Weight
For determination of the intrinsic viscosity, it is necessary to extrapolate the hsp/c vs c plot to infinite dilution (c ® 0), since it is only under this condition that the polymer molecules in solution contribute to viscosity discretely without mutual interference. Solubilization of a polymer sample is preceded by a large amount of swelling if left undisturbed and the degree of swelling is higher in a better solvent. Likewise, the intrinsic viscosity is also higher in a good solvent than in a poor solvent. What it really means is that in a better solvent, as the polymer goes into solution, a unit mass of polymer expands more to give a higher hydrodynamic volume.
Let us now consider a heterogeneous polymer in dilute solution of concentration c behaving ideally in that the individual molecules contribute to viscosity independently of one another. In that event, if (hSP) is the specific viscosity contribution due to species of size i, then one may write:

clearly, for a approaching the value of unity, v approaches w and in the limiting case of a = 1, v = w.
The viscometric studies of polymer solutions as a means of molecular characterization of polymers are well recognized and widely practised because of simplicity in terms of experimental approach and the apparatus needed. Dilute solution viscosity is conveniently measured in capillary viscometers of different kinds such as the Ostwald type or the Ubbelohde type. Ubbelohde viscometer is a suspended level viscometer and it has the advantage that the flow time measurements are not dependent on the volume of liquid (solution or solvent) in the viscometer and hence, measurements at a series of concentrations can be conveniently made by successive dilution within the viscometer. All flow time measurements for the solvent and solutions of different concentrations are made in a thermostat bath regulated within ±0.1°C. The flow time data should then be treated graphically according to Eq. (19) or (20) and then extrapolated to infinite dilution (c® 0) to obtain the value of the intrinsic viscosity, [h] as described earlier. v can then be evaluated using the Mark-Houwink equation and using the appropriate K and a values from the literature, if available, or from an independent determination as described earlier in this section.
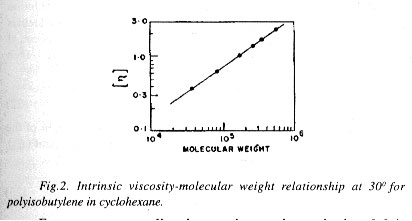
Plastics: Materials and Processing Technology
1. Plastics Materials - Introduction
Material systems which can be conveniently made to flow or deform into a desired shape or form, usually under heat and/or pressure, are commonly known as plastics. The binding material in a given plastic product is invariably a polymer or resin. A resin is defined as a material, organic in nature and of either synthetic or natural origin, which has the capacity to produce a thin coherent film either from its melt or from its solution. A resin may be polymeric (characterized by high molecular weight and a characteristic repeat (chemical) unit in the structure) or non-polymeric (characterized by low molecular weight) in nature. According to the resin-concept as above, almost all polymers may be considered as resins, but all resins are not polymers. Conventionally, however, resins are brittle materials showing different degrees of hardness under ambient conditions. Rosin and Shellac are two important natural resins, and among the relatively low molecular weight (M [ 2000) synthetic resins, the coumarone-indene resins and the so called petroleum resins are important. These resins and their derivatives find widespread use as tackifiers and as improvers of adhesion, gloss, hardness, etc., in many adhesive and coating formulations based on different polymers. Most commercially important structural resins are, however, thermosetting polymers covering such products as the phenolic (phenol-formaldehyde) resins, amino (urea-formaldehyde and melamine-formal-dehyde) resins, alkyd (polyester) resins, epoxy resins, etc.[/P]
The technology of a polymer begins with the polymer itself, which is the base material of a plastic, rubber, adhesive, coating or related item that we see around us. The synthetic polymer industry can be either coal-based or petroleum-based. Ethylene, an important monomer and an intermediate for some other monomers may also be obtained by dehydration of alcohol. The developing trends in the decades after 1950 switched the technology of monomer synthesis for almost all kinds of polymer in favour of petroleum as the basic raw material, so much so that today's monomers and polymers are widely recognized as petrochemicals. Salient features of polymerization relating to some important polymers finding plastic applications in the main and their properties, processing technology and applications will be discussed in the present chapter.
2. Polyethylene
Polyethylene of different density ranges can be obtained by chain polymerization of ethylene, the simplest olefinic monomer. The observed chemical nature and physical properties of the polymer depend largely on the nature of the catalyst used and conditions employed during polymerization. Polyethylenes obtained by polymerization of ethylene are mostly branched polymers, the chemical nature and frequency of branches being largely dependent on reaction conditions and the mechanism involved. Polyethylenes of commerce are available in two major density ranges and the classified products are commonly known as: (a) low density polyethylene, LDPE (density range, 0.91 5-0.94 g/cm3), and (b) high density polyethylene, HDPE (density range, 0.945-0.96 g/cm3).
Low Density Polyethylene (LDPE)
LDPE is produced by free radical polymerization of ethylene under very high pressure (1500-3000 atm), using oxygen, peroxides or azocompounds as initiators. The development of a commercial process of this kind was associated with numerous technical difficulties. Highly exothermic nature of the polymerization requires strict control of reaction variables (pressure, catalyst concentration, temperature, monomer purity, flow velocity in the reactor, etc.) in order to ensure safety and to prevent run-away reactions and explosion. The early ICI process, commercialized in 1939, employed oxygen (air) as the catalyst. The polymerization reaction, simply, written as is expected to be favoured under high pressures and low temperatures from thermodynamic considerations. On kinetic considerations, however, a temperature range of 170-250°C is usually employed. At a lower temperature, the radical generation process is far too slow for commercial production of the polymer.
nCH2 = CH2 ® - (CH2 - CH2)n-
To get high molecular weight, ethylene of high purity is required. Traces of oxygen catalyzes the polymerization process. The allowable oxygen at a pressure of 2000 atm and 165°C is about 0.075%. If this limit is crossed, explosive decomposition reaction occurs yielding hydrogen, methane and carbon in the main. The conditions are so chosen as to permit and favour only polymer formation at commercially viable rates. High pressure increases local concentration of the gaseous monomer in the vicinity of the transient chain radicals, and thereby brightens the prospect of obtaining high molecular weight for the polymer product. Higher temperature, however, tends to lower the molecular weight.
A pressure of the range of 1500-2000 atm is obtained by compressing refined ethylene, usually in two or three successive stages. For continuous operation, the reactants are passed through narrow-bore tubular reactors or stirred reactors, fitted with efficient heat-transfer system. Conversion in the range of 10-30% is normally achieved after which the unreacted monomer is flashed out and recovered and the viscous molten polymer is extruded out in the form of strands or ribbons, cooled and granulated.
In the conventional activation with oxygen, the formation of some unknown peroxide/hydroperoxide must be considered as the source that furnishes the radicals for chain initiation on subsequent decomposition, which apparently takes place fast enough only above 150°C. The heat of polymerization of ethylene1 (800-1000 cal/g) is considerably higher than that of other monomers, e.g., styrene (164 cal/g). In addition to many difficulties inherent in high pressure reactions, the control of temperature consequent to this high heat of polymerization is one of the most important problems in the polymerization of ethylene by the high pressure technique.
Chain branching (frequency, branch-length and distribution) as well as molecular weight and molecular weight distribution can be conveniently varied and controlled by varying the temperature, pressure, nature of initiator and its concentration and other additives such as solvents/non-solvents or suitable chain transfer agents, and by injecting the initiators at different points in the reactor.
High Density Polyethylene (HDPE)
Use of coordination catalysts or supported metal oxide catalysts allows polymerization of ethylene at a much lower pressure as well as at a relatively low temperature; the polymers thus produced are by and large linear, having much fewer branches than found in the product obtained by the high pressure free radical process. The HDPE synthesis is conveniently done by (i) the Ziegler process using an aluminium trialkyl-TiCl4 complex as the catalyst dispersed in a hydrocarbon solvent such as n-heptane and employing a slightly elevated pressure and a temperature in the range of 70-100°C, (ii) the Phillips process, using CrO3 (5%) supported on finely divided silica-alumina (75-90% silica) base as the catalyst. The catalyst is activated by heating to about 250°C, dispersed in a hydrocarbon solvent such as cyclohexane and polymerization of ethylene is done employing a pressure of 15-30 atm and a temperature in the range of 130-150oC, or (iii) the Standard Oil (Indiana) process using a dispersion of molybdenum oxide catalyst containing sodium and calcium metal or their hydrides as promoters in a hydrocarbon solvent, employing a pressure in the range of 49-80 atm and a temperature range of 220-250°C. The Ziegler process yields polymers of density of about 0.945 g/cm3 while the other processes using (supported) metal oxide catalysts yield polymers of density as high as 0.96 g/cm3. On completion of reaction in the Ziegler process, the catalyst is destroyed using water or alcohol with traces of an acid and the polymer is collected by nitration or centrifugation, which is then washed and dried. The processes using metal oxide catalysts may be either fixed-bed, moving-bed or slurry processes. On completion of the reaction, the polymer formed and evenly distributed in the paraffin or cycloparaffin diluent in the high reaction temperature is separated from the catalyst fragments and then recovered and isolated by cooling and/or solvent evaporation.
Structure and Properties of Polyethylenes
The differences in the density range of LDPE and HDPE as mentioned earlier have their origin in the inherent difference in the mechanistics which are consequential in giving rise to significant structural differences in these polymers. The structural differences relate to differences in molecular weight and molecular weight distribution and differences in degrees of chain branching in the main. Due to the high chain transfer in the radical polymerization effected under high pressure, the resultant polymer, LDPE has nearly 20-50 branches (both long and short branches) per 1000 linear carbon atoms in the chain molecules. The loss of molecular symmetry due to the high degree of branching results in lower density range (0.915-0.94 g/cm1, and lower softening or melting temperature (110-117°C) for LDPE. The low pressure polymers, HDPE, are by and large linear, having only 2-5 short branches or side chains per 1000 carbon atoms in the main chain, thus having a higher density range (0.945-0.96 g/cm3) and higher melting temperature (125-130°C) compared to LDPE. The theoretical linear (unbranched polymer N polymethylene,-(CH2)n-as obtained from diazomethane using BF3 - Et2O catalyst at about 0°C through elimination of N2 on polymerization [nCH2N2 ® - (CH2)n - + nN2] is, however, characterized by high molecular weight with a density as high as 0.98 g/cm3 and a melting temperature of 137°C.
Polyethylenes and other polyolefins are graded by what is known as the melt flow index (MFI), i.e., the weight in grams extruded by piston action through a standard opening under a standard load in a standard plastimeter at 190°C over a specified time period (see also Chapter 10, MFI gives a rough guide to processability of different grades of polymers of a given kind or monomeric origin. For a given kind of polymer a higher MFI indicates lower molecular weight and vice versa. LDPE comes in a wide MFI range (0.3-70), but for HDPE, the useful MFI range is much lower (0.02-2.5). The tensile strength of LDPE ranges between 1500-2500 psi. and that for HDPE is in the range of 3000-5000 psi. The Izoc impact strength for LDPE is as high as 10 ft lb per inch of notch, white that for HDPE is much lower (1.5-5 ft lb per inch of notch).
Polyethylenes are chemically paraffinic in nature, inert to non-oxidizing acids, alkalies, water and many aqueous solutions and organic solvents HDPE is usually more resistant to chemicals than LDPE. There is no singular solvent for polyethylenes at room temperature. LDPE dissolves in toiuene at or above 60°C, while HDPE dissolves at temperatures about 20-30°C higher in the same solvent. With larger amorphous regions, LDPE is more permeable to gases than HDPE. Both LDPE and HDPE are prone to aerial oxidation, which is quite severe at processing temperatures. LDPE is more prone to oxidation than HDPE (if free from metallic impurities) in view of higher concentrations of tertiary hydrogen (at branch sites) in LDPE. Uncontrolled oxidation leads to measurable incorporation of carbonyl and peroxy groups which make the polymer more prone to degradation and embrittlment due to absorption of heat and UV light. Polyethylenes generally show poor environmental stress cracking resistance, LDPE being poorer in this respect than HDPE. However, improved processing conditions and better design result in low stresses being imparted to products, thus ensuring desired long service life and performance for them. The stress cracking problem is less critical with relatively high molecular weight (relatively low MFI) and high density materials.
Polyethylenes are very good insulating materials and in this respect they compare favourably with other dielectric materials. Being adequately flexible, LDPE is extensively used as an insulator in wires and cables.
Uses and Applications of Polyethylenes
Polyethylenes, being thermoplastic in nature, are conveniently processed by injection moulding, blow moulding, rotational moulding, extrusion and thermoforming techniques. They find extensive uses and applications as moulded or formed objects, films, sheets, bottles and containers, pipes and tubes, and in wire insulation and cables. Films, commonly made from LDPE, find application in packaging, water proofing, irrigation and water management including canal lining and mulching, and in coating and lamination. Various additives such as fillers, pigments and colouring matters, antioxidants, slip additives, anti-block and antistatic agents, UV-absorbers, blowing agents and flame retardants are mixed or blended with polyethylene, polypropylene and other polyolefins for various reasons. For high voltage cables application and for heat resistant films or moulded objects, polyethylene is preferably cross-linked using selected chemical cross-linking agents, such as peroxides or by irradiation with high speed electrons or g-rays. Factors contributing to widespread application of polyethylenes are low cost, easy processability by variety of techniques, excellent chemical inertness and electrical insulation properties, low temperature toughness and flexibility, good clarity of thin films, heat sealability, non-toxic nature and low water vapour permeability. The major limiting factors in this respect are low softening point or heat distortion temperature, proneness to environmental stress cracking, poor scratch resistance, low tensile strength and high gas permeability.
Chlorosulphonated Polyethylene
The high molecular symmetry of polyethylene (LDPE) may be broken by treating the polymer with chlorine in presence of small quantity of sulphur dioxide, thereby introducing some - SO2Cl groups and-C1 atoms along the polymer chain as shown in the reaction scheme

The chlorosulphonated polyethylene (CSP) rubbers are superior to many hydrocarbon (diene) rubbers in ozone resistance. They are also among the best chemical-resistant rubbers and have good resistance to heat-aging and flexing.
Chlorination of ethylene to different degrees (up to about 40% chlorine) is also practised to obtain different products-thermoplastic or elastomeric. The reaction is carried out at an elevated temperature in solution or in suspension using either LDPE or HDPE. With HDPE, chlorination in aqueous suspension at or above 120°C gives a convenient elastomer that can be vulcanized by peroxides.
3. Linear Low Density Polyethylene (LLDPE)
The understanding of a link between different kinds and degrees of branching and different ranges of densities as exemplified by LDPE and HDPE has led to the more recent development of a mechanistically linear "polyethylene of a low density range by a copolymerization approach. The product, a linear copolymer of ethylene and a minor proportion of an a-olefin such as l-butene, l-hexene or l-octene, appears as a branched polyethylene having controlled proportions of uniform short chain branching along the chain. Uniform short branches appear in the chain at each point of the a-olefin comonomer incorporation. This linear grade of polyethylene, having a low density range (0.915-0.934 g/cm3) is commonly known as Linear Low Density Polyethylene (LLDPE) and it can be synthesized employing the Ziegler process or the Phillips process.
Structurally, LDPE and HDPE may also be considered, as copolymers of ethylene and minor proportions of a-olefins, in view of the presence of significant long and short branches in LDPE and very limited short branches in HDPE. While LDPE and HDPE, if so considered as copolymers, are copolymers by incidence and not by definition or design. LLDPE has the distinction of being a copolymer primarily by definition and design.
LLDPE displays a superior balance of toughness, stiffness, stress cracking resistance and electrical properties over LDPE. With favourable production economics and progressive acceptability in the market, LLDPE may soon take over up to 30-50% of the traditional LDPE market throughout the world.
There are significant differences between the rheological behaviours of LDPE and LLDPE which pose some resistance towards acceptance of LLDPE by many end product manufacturers. It is, however, most likely that on grounds of superior performance, most non-film applications of LDPE will finally switch over to LLDPE. Judicial blends of LDPE and LLDPE are also being developed for various applications. The melting points of commercial grades of LLDPE are in between the melting points of commercial LDPE and HDPE.
4. Polypropylene
The polypropylene of commerce is isotactic polypropylene. Its synthesis became possible through the commercial utilization of coordination polymerization during 1957-60. Propylene, the monomer, is obtained from the cracking of petroleum products (natural gas, light oil or naphtha). The separation of propylene from propane in the C3 fraction is difficult and is achieved by fractional distillation in a carefully designed distillation plant. Propylene of high purity is required for polymer synthesis.
Synthesis of Polypropylene
The synthetic approach for polypropylene is much the same as that for the production of HDPE by the Ziegler process. The same reactor can be used to polymerize ethylene, propylene or other a-olefins with some modifications, if necessary. For propylene, the reaction condition should be so chosen as to overwhelmingly favour formation of the isotactic polymer. The catalyst may be prepared from TiCl4 and aluminium triethyl combination dispersed in naphtha. Propylene is charged into the reactor under pressure. Nearly 80-85% conversion is achieved in 8-10 h at 60°C. The reaction variables are molar ratio of catalyst components, catalyst concentration, reaction temperature and monomer pressure. Hydrogen may be used as achain terminating or transfer agent for control of molecular weight. About 8-15% atactic polymer is also formed along with the formation of the isotactic polymer in the main. After the reaction reaches 80-85% conversion stage, the ingredients are transferred into a flash drum to purge off the unreacted monomer which is then recycled. The atactic product is soluble in the solvent naphtha. The solution is decanted off after centrifugation whereby most of the atactic material is removed. The residue is then treated with methanol acidified with traces of HCl to decompose and dissolve the catalyst. The polymer is then centrifuged washed, dried and collected.
Structure and Properties of Polypropylene
Polypropylene (PP) is the lightest homopolymer known, its density being 0.90 g/cm3. With a rigid, short methyl group attached to every alternate carbon atom in the linear molecular chains and the highest order of steric regularity, (isotactic) polypropylene, as commercially produced, is comparatively more rigid, stiff and strong than HDPE. Its melting point is nearly 45-50°C higher than the melting point of HDPE. Having a much higher concentration of tertiary carbon atoms in its chains, PP is much more prone to oxidation or aging than HDPE.
Commercial PP is nearly 90-95% isotactic. The degree of isotacticity or the isotactic index is given by the percentage of the polymer that is insoluble in n-heptane. Isotactic PP is much less crystalline than HDPE. The high melting point (³ 170°C) of PP allows the moulded objects to be steam sterilized. Its high strength (tensile strength: 4500-5500 psi, elongation at break 100-300%), high melting point and low density is advantageous in many applications. PP, however, becomes brittle close to 0°C. In this respect, PP is inferior to polyethylene. But PP has a much superior hinge property. The problem of brittleness is very well overcome by block copolymerization of propylene with minor proportions (5-15%) of ethylene. PP is generally free from environmental stress cracking problems. Its specific heat is lower than that of polyethylene but higher than that of polystyrene. PP melt is more non-Newtonian than polyethylene melt. The melt viscosity of PP is also more sensitive to change of temperature. It is also graded by melt flow index for processing purposes. Generally, PP shows lower moulding shrinkage than polyethylene. If free from catalyst fragments, PP and polyethylene are similar in electrical properties.
PP exhibits higher clarity (less opacity) than HDPE, mainly because the amorphous and crystal densities are closer for PP than for polyethylene. Biaxially oriented polypropylene (BOPP) shows a high degree of clarity and biaxial stretching also produces polymers of much improved tensile strength. PP is used in the form of moulded and formed objects, sheets and films, bristles, monofilaments and fibres, covering such items as luggage, frames, containers and different packaging items, ropes, tows and nets, pipes and tubes, etc. Typical mouldings also include components of automobile and home appliances industry, machine parts and components, sterilizable health-care and hospital equipment, kitchenware; furniture, etc.
5. Copolymers of Ethylene
Among the copolymers of ethylene, those between ethylene and propylene and between ethylene and vinyl acetate have reached a stage of widespread development and application. Other important copolymers involving ethylene and some other a-olefin such as l-butene, include the wide range of linear low density polyethylene (LLDPE). The a-olefin copolymers are produced by the Ziegler or Phillips process, while copolymers of ethylene with vinyl acetate, maleic anhydride and acrylate monomers are industrially produced by radical polymerization.
Stereo block copolymers of propylene and ethylene (4-15% ethylene) show improved impact properties. Though copolymeric in nature, they are essentially considered as polypropylenes of minor chemical modifications. When the two monomers are used in nearly equal or 60 : 40 proportions, products commonly known as ethylene-propylene rubbers (EP rubbers) are formed on copolymerization. EP rubbers are more aging resistant than diene rubbers, being devoid of unsaturations and they are conveniently vulcanized by peroxides, e.g., dicumyl peroxide. Carbon black is used with EP rubber to develop moderate to high tensile strength and modulus. Low dose of sulphur (0.3 phr.) is often used along with a peroxide (2-3 phr.) as the curative and Ca-stearate is preferred to stearic acid as the activator.
Ethylene vinyl acetate (EVA) copolymers are available in different grades varying in vinyl acetate (VA) content (3-48%). Those containing low VA content (3-5%), are considered as modified low density polyethylenes. Their high surface tack, disadvantageous in conventional film applications, is taken advantage of in meat packaging as stretch films as well as for cling-wrap purposes. In higher vinyl acetate compositions, EVA copolymers are used as wax additives, wax coatings and hot melt adhesives. Selected grades of hydrolyzed EVA, commonly designated as EVOH, offer excellent barrier properties with respect to gas (oxygen) permeability.
Copolymerization of ethylene with carboxylic monomers such as maleic anhydride, methacrylic acid, etc., by the high pressure process produces materials which are used as thermosetting materials but processed like thermoplastics. When treated with metal salts, oxides or alkoxides (Zn, Ca, Mg, etc), ionic (metal carboxylate) cross-links, stable under ambient conditions, are readily established. The ionic cross-links get weakened with rise in temperature and their influence disappears at the processing (melting) temperatures, thus permitting reshaping. On cooling after reshaping, the ionic cross-links are regrouped and reformed and the thermosetting character reappears. Such polymers with "meltable" cross-links are known as "ionomers".
6. Polystyrene
Monomer Synthesis
Polystyrene is another high tonnage, general purpose thermoplastic polymer. The monomer, styrene, or vinyl benzene, CH2 = CH.C6H5 is made from benzene and ethylene in two steps. The first step involves alkylation of benzene (formation of ethyl benzene) through reaction with ethylene in the presence of a Friedel-Crafts Catalyst such as AlCl3.
In a subsequent step, ethyl benzene is dehydrogenated to styrene by heating its stream over iron oxide or magnesium oxide catalyst at about 600°C. Styrene, isolated from the dehydrogenation product by distillation, is stabilized by addition of traces of a phenolic stabilizer, such as t-butyl catechol and then stored in a cold storage for longer shelf-life.
Polymerization of Styrene
Polymerization of styrene is mostly done following the bulk polymerization or suspension polymerization techniques. The bulk polymerization is done in two stages. In the first stage, the stabilizer-free monomer containing dissolved initiator is allowed to polymerize at about 80°C in one or more "prepolymerizer" batch reactors, each fitted with an efficient stirrer and an outer jacket for circulation of heat transfer fluid until a conversion of about 30% is reached. The viscous monomer-polymer mixture is then forced into the top of a tower reactor (nearly 25-40 ft in height and 10-15 ft in diameter) which is fitted with heating and cooling jackets and coils for external and internal control of temperatures respectively. The temperature down the tower changes from 100°C at the top to nearly 180-200°C at the bottom. The melt containing polystyrene and traces of the monomer passes through an extruder at the bottom from which it emerges as narrow strands or filaments which are then cooled, granulated and packed.
Structure and Properties of Polystyrene
Commercial polystyrene is a radically initiated linear atactic polymer. It is amorphous and transparent. It comes in different grades such as: general purpose grade, high molecular weight grade and heat resistant grade. Removal of last traces of unreacted monomer (from 5-0%) improves the softening point or heat distortion temperature from nearly 70°C to about 100°C. The polymer is hard, brittle and transparent at room temperature. It is soluble in aromatic hydrocarbon solvents, cyclohexane and chlorinated hydrocarbons. The polymer has a density of 1.054g/cm3, a refractive index of 1.59-1.60 and a Tm value of 240°C. It sounds like metal when dropped. It decomposes at elevated temperature into a mixture of volatiles, a significant part of which is the monomer which can be identified from its characteristic smell.
Polystyrene, being hydrocarbon in nature, has very low moisture absorption; this feature coupled with its good mouldability, dimensional stability and low moulding shrinkage makes polystyrene an ideal polymer for injection moulding and for thermoforming. As a rigid thermoplastic, it has moderate tensile strength (6000-8000 psi) and very low elongation at break (1-3%). The major limitations of the polymer are its brittleness and very low impact strength (Izod 0.2-0.35 ft lb in-1 notch) and low heat distortion temperature (~80°C).
Polystyrene has poor outdoor weathering resistance and a tendency to yellowing and crazing on long use. It possesses very good electrical insulation characteristics and a low dielectric loss factor at moderate frequencies.
Modification to High Impact Grades
There have been many attempts to improve impact properties of polystyrene, but those achieved through use of rubbery additives and through copolymerization approach have been commercially successful and widely practised. Rubber modified polystyrenes are used to nearly the same extent as straight polystyrene. Rubbers commonly in use for the purpose are styrene-butadiene rubber (25-30% styrene content) or simply polybutadiene rubber. Simple blending of the rubbery polymer with polystyrene done on two - roll mills or in the latex from brings only marginal improvements in impact strength. Much better improvements are achieved by polymerization of styrene in an emulsion of the rubbery polymer or in solution containing the rubbery polymer, through what may be termed as a graft copolymerization approach. To derive the best effect, the rubber used should be semicompatible with polystyrene and should have limited degrees of cross-linking. The rubber should be dispersed in fine discrete droplets within the polystyrene matrix, the rubbery phase comprising only 5-20% of the total volume of the resin-rubber composite. Such high impact grade polystyrenes may have two to five times the impact resistance of straight polystyrene, but they exhibit nearly half the tensile strength and a much lower softening point. They also show inferior aging resistance and require stabilization through incorporation of adequate doses of antioxidant stabilizers.
Styrene-Acrylonitrile (SAN) Copolymers and ABS Resins
To obtain a styrene-based polymer of higher impact strength and higher heat distortion temperature at the same time, styrene is copolymerized with 20-30% acrylonitrile. Such copolymers have better chemical and solvent resistance, and much better resistance to stress cracking and crazing while retaining the transparency of the homopolymer at the same time. In many respects SAN copolymers are also better than poly (methyl methacrylate) and cellulose acetate, two other transparent thermoplastics.
ABS resins are terpolymers of acrylonitrile, butadiene and styrene, prepared by interpolymerization (grafting) of styrene and acrylonitrile on polybutadiene or through blending of SAN copolymers with butadiene-acrylonitrile (Nitrile) rubber. Impact improvement is far better if the rubber in the blend is lightly cross-linked. The impact resistance of ABS resins may be as high as 6-7 ft lb. per inch of notch.
Processing, Uses and Applications of Polystyrene
Polystyrene and related copolymers, terpolymers and blends are widely processed by injection moulding, blow moulding, thermoforming and extrusion techniques. Expanded polystyrene finds extensive use in packaging and shock absorbing applications, in thermal insulation and as acoustic improvers in halls and auditoria. Largest outlet of polystyrenes is in the packaging field. High impact grades are suitable for use as toys, games and sports articles, casings and cabinets for electrical/electronic gadgets and equipments and inner liners of refrigerators. ABS resins have emerged as engineering plastics for toughness, strength and durability and are being increasingly used as housewares and as housings for domestic and industrial electrical equipment, automobile components, etc. Another major outlet of polystyrene is in the making of ion-exchange resins. For this, the polymer is produced in the cross-linked form by suspension polymerization technique using divinyl benzene (1-5%) as the comonomer. Modifications of the suspension resin beads by controlled sulphonation with concentrated H2SO4 produces cation-exchange resin. Chloromethylation of the resins with chloromethyl ether and their subrequent transformation into quaternary ammonium salts by treatment with tertiary amines followed by alkali treatment lead to the formation of anion-exchange resins.
7. Acrylic Plastics: Poly (Methyl Methacrylate)
Plastics based on acrylate and methacrylate monomers are commonly known as acrylic plastics. Most important among the acrylic plastics is poly(methyl methacrylate) prepared by the polymerization of methyl methacrylate. Monomer methyl methacrylate is prepared from acetone through the cyanohydrin route. The cyanohydrin is treated with 98% sulphuric acid in a cooled kettle to yield methacrylamide sulphate intermediate which on esterification with methanol produces methyl methacrylate. The synthetic route is shown in the reaction scheme (5).

Moulding grade poly (methyl methacrylate) is made by bulk or suspension polymerization of the monomer, using peroxide or azonitrile initiators at about 100°C, preferably in the absence of air. For the production of cast sheets, the monomer is first converted into a prepolymer syrup of convenient viscosity and the syrup is then poured into the casting cell consisting of heat resistant, parallel, polished glass plates separated by rubber gaskets round the edges, and held together by spring loaded clamps or spring clips. Precautions must be taken to obtain sheets free of sink marks and voids. The cell is then gradually heated in a heating tunnel for 15-20h over a temperature zone of 50-100°C to complete the polymerization. Rods are made by polymerizing the syrup under pressure in vertical aluminium tubes, while tubes are prepared by polymerizing a calculated amount of syrup in an aluminium tube under nitrogen pressure, with its both ends closed. The assembly is heated as the tube is rotated horizontally at a constant rate, and the syrup polymerizes on the wall of the rotating tube. Natural shrinkage on polymerization enables easy removal of the casting.
A combination of excellent optical clarity (refractive index, 1.49), total lack of colour, some toughness and outstanding outdoor weathering behaviour makes poly(methyl methacrylate) very useful as automotive tail lamp and signal light lenses, jewelry, lenses of optical equipment and contact lenses. The polymer is very popular in display and advertisement applications; use in the building industry is also notable.
Minor proportions (5-25%) of different monomers such as n-butyl acrylate, 2-ethyl hexyl acrylate, methacrylamide, dimethyl aminoethyl acrylate, etc., are used to make coating grade poly(melhyl methacrylate), usually by solution or emulsion techniques, for paints and enamel applications. Production of dentures is another important outlet of poly(methyl methacrylate). Poly(methyl acrylate) and poly(methyl methacrylate) may be saponified to form the polymeric acids which correspond to polymers of acrylic acid and methacrylic acid. They show characteristic/polyelectrolytic behaviour in aqueous solution. They are not suitable for use as formed or moulded objects, films, sheets, etc. As such or as sodium salts they are useful as thickening agents.
8. Acrylic Fibres
Fibres primarily based on polyacrylonitrile are known as acrylic fibres. Acrylonitrile monomer is produced by addition of HCN to acetylene or by dehydration of ethylene cyanohydrin formed by the addition of HCN to ethylene oxide. Acrylonitrile is a monomer which is a non-solvent for its polymer. Bulk polymerization turns out to be a case of heterogeneous polymerization and is associated with high heat generation and consequent to that, with hazards of violent explosion. The monomer is soluble in water to the extent of nearly 8% at room temperature and polyacrylonitrile of commerce is conveniently formed by heterogeneous aqueous polymerization of acrylonitrile using redox catalysts at or near room temperature. The polymer is obtained in the form of a fine precipitate.
Polyacrylonitrile, i.e., the homopolymer of acrylonitrile is unsuitable for many conventional thermoplastic applications as it suffers discolouration and infusibility on heating. It starts yellowing on heating and finally turns black, insoluble and infusible. It is ordinarily insoluble in most common solvents. Dimethyl formamide, dimethyl sulphoxide and tetramethylene sulphone are good solvents for the polymer, however. It also dissolves in aqueous concentrated solution of some salts. The polymer is dissolved in one of these solvents and then processed by wet or dry spinning techniques to produce the fibre.
To improve certain properties such as dyeability, feel and texture of the acrylic fibre, the polymer is prepared by copolymerizing 5-15% of one or more monomers, such as vinyl pyrrolidone, vinyl acetate, acrylates, acrylic/methacrylic acid, along with acrylonitrile. The incorporation of the comonomer units makes the fibre more suited for dyeing with many common dyes for textiles. Fibres from copolymers having much higher proportions (20-35%) of a comonomer, usually vinyl chloride and vinylidene chloride, are commonly known as modacrylic fibres.
The commercial acrylic fibres come in different colours and shades. They have outstanding weathering resistance. They also exhibit good resistance to chemicals, stains, insects and microorganisms. In bulkiness, feel and warmth, the acrylic fibres are very much similar to wool. They are widely blended with other fibres, particularly wool, to form various textile items.
