Modified Natural Products
CUPRAMMONIUM RAYON
In 1857, the German chemist and botanist, Schweitzer, discovered that cotton and paper were soluble in an ammoniacal copper hydroxide solution Cu(NH3)4(OH)2]. Then, later in the nineteenth century, the American inventor, Weston, produced filaments from such a solution for electric lamp filaments. In 1890, the French chemist, Despaissis, was granted the first patent for making a cuprammonium artificial silk. The first commercial development took place in Germany under Pauly's patent but was later abandoned. Much later, in 1918-19, a German firm, Bemberg A.G., started production, using a different process, and several plants in Europe produced this type of rayon. It was not until 1924-25 that an American plant, established by Bemberg, began production in this country.
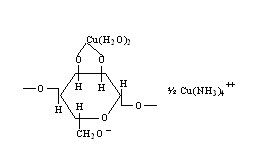
The principal raw materials used in the cuprammonium process are purified cotton linters or wood pulp, the latter being more generally used today. In one process, aqueous copper carbonate is mixed with the cellulosic material in a shredder to form copper cellulose. After expression of liquid, the residual cake is mixed with aqueous ammonia and caustic soda to form a viscous solution. The solution, containing about 8 per cent cellulose, is stored, filtered, and vacuum deaerated. Glucose is usually added to stabilize the solution and improve spinning properties. The cellulose now probably exists in the form represented by where the original ammonia-copper complex is destroyed and two molecules of water are complexed with the copper.
The spinning solution is forced through relatively large holes in nickel spinnerets; subsequent stretching of the filaments reduces their diameter. A thread from a 0.0002 inch orifice may be stretched to as small as one denier in this process. As the filaments emerge from the spinnerets, they pass through coagulating baths, which may be acid or alkaline. Sulfuric acid baths are now less common than alkaline baths. The yarn is then washed, dried, and wound on bobbins. As many as 400 threads are spun simultaneously. After treatment with dilute sulfuric acid to remove residual copper, the yarn is bleached, washed again, and dried.
It is essential that the copper salts and the ammonia used be recovered, else the cost of the process would be prohibitive. In this sense, the production of cuprammonium rayon parallels that of soda ash by the ammonia process, wherein the intermediates are of greater value than the final products, and the process is competitive only because of an efficient recovery process. The ammonia is recovered from the dissolving tank and from alkaline coagulating tanks by passage through an ammonia scrubber. Copper from coagulating baths and from the acid and water purification systems is recovered by precipitation. Approximately 0.57 pound of ammonia, 0.40 pound of caustic, 0.75 pound of copper salt, and 0.92 pound of cotton linters are needed to make one pound of cuprammonium rayon.
Cuprammonium rayon (often referred to as Bemberg rayon because the Bemberg Corporation is probably the sole producer) resembles real silk in fineness of filaments, luster, and softness. However, since it is regenerated cellulose, it also has most of the properties of cotton and viscose rayon. Probably its chief virtue is its capability of being drawn to very fine filaments, as low as 0.4 denier. The fineness of filament gives cuprammonium rayon great pliability and good draping qualities; hence it is used in apparel manufacture, especially for women's wear. It has been widely used as a substitute for silk.
Due to the inherently high cost of manufacture, even with a good recovery process, the production of cuprammonium rayon is very small. In the United States today, probably no more than 3 per cent of the rayon is manufactured by this process, the remainder being made by the viscose process.
VISCOSE RAYON
Although the viscose process for making rayon was the last of the three commercial processes to be developed, it has, since about 1910-11, been by far the most important. The process is rather lengthy (taking several days to a week) and requires careful control throughout; yet all the raw materials are fairly cheap, with the result that viscose rayon can generally be made to sell below the price of all other rayons. This is the chief reason for its greater success.
The development of viscose rayon is due mainly to the work of two English chemists, Cross and Bevan, who obtained a patent in 1895 on rayon manufacture. They discovered that when cellulose is treated with caustic soda and then with carbon disulfide, it is converted into a new compound. This new compound, when dissolved in caustic soda, produces a viscous solution which Cross and Bevan termed "viscose."
As with cuprammonium rayon, cotton linters or wood pulp are used, the latter being preferred due to its lower cost. [A viscose plant has been built in the Philippines recently to covert sugar-cane waste (bagasse) into rayon]. Cellulose pulp from cotton linters, northern spruce, western hemlock, or southern pine is steeped or mercerized in an 18 per cent caustic soda solution to form sodium cellulose. After expression of liquid in hydraulic presses, the sodium cellulose is shredded into crumbs. The crumbs are aged for two to three days in steel or iron cans so that partial oxidation and depolymerization occur. They are then blended with carbon disulfide vapor in large, rotating churns under carefully specified and controlled conditions. A gelatinous mass of sodium cellulose xanthate is formed [the sodium salt of the cellulose acid ester of xanthogenic (dithiocarbonic) acid, HO.CSSH], together with some adsorbed caustic soda:
[C6H7O2(OH3)]n + NaOH + CS2 ] [C6H7O2(OH)2.O.CSSNa]n + H2O
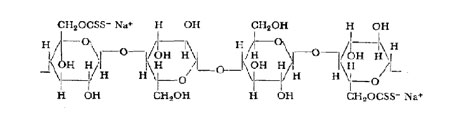
Although almost complete xanthation can be obtained, normally only about one to two hydroxyl groups per cellobiose residue are xanthated, as this is sufficient to allow solution or dispersion of the product in more caustic solution.
A portion of a cellulose xanthate chain may be represented as follows:
The solubility of this polymer in aqueous media is due to its lack of symmetry, which decreases hydrogen bonding, and to the ionic character of the xanthate group. Thus, a dilute caustic solution is added to the xanthate in a mixer, and the mass is stirred until a homogeneous, viscous mass of viscose is obtained. At this time, the luster of the rayon is controlled. If nothing is added, a high luster rayon is obtained; to obtain a dull or semidull yarn, a pigment, usually titanium dioxide or a mineral oil, is added which causes the yarn to reflect diffusely. Since these additives are incorporated into the yarn, they cannot be later washed out.
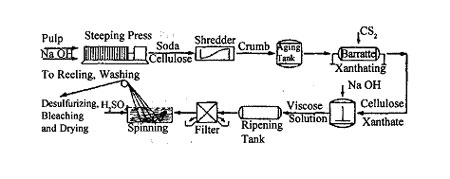
During the subsequent ripening period, which takes from four to five days and is done in tanks in an insulated room at 15° to 20° C, the molecular structure is retained, but the viscosity slowly decreases, then rises again, partly due to regeneration of some cellulose by slow hydrolysis of xanthate and partly by the salting-out of inoganic by-products. The regenerated cellulose is maintained in emulsion form by the undecomposed xanthate, which acts as a protective colloid. During this process, the color of the mass turns from whilte to an orange-yellow.
After proper ripening, the solution is filtered, vaccum deaerated, refiltered, and spun, as with cuprammonium rayon. The spinneret may be a platinum alloy containing from 10 to 120 holes for ordinary rayon and more than 3,000 for tire cord and tow. The holes are generally from 0.002 to 0.004 inch in diameter. The final size of the filaments depends more on the rate of supply of the solution to the spinneret and the amount of stretching during coagulation than upon the size of the spinneret holes. All of the monofilaments from a single spinneret are combined into a single thread of rayon yarn.
The spinning bath, made of sheet lead, contains a solution of sulfuric acid and additives maintained at 40° to 55°C. A representative composition is as follows:
| |
Per Cent |
| Sulfuric acid |
9-12 |
| Sodium sulfate |
17-20 |
| Glucose |
2-5 |
| Zinc sulfate |
1 |
| Water |
67-71 |
Sometimes a small quantity of magnesium sulfate is also added.
Although the composition of the bath has been determined empirically, the probable functions of the ingredients are as follows: The sodium sulfate precipitates the sodium cellulose xanthate from the viscose solution in the form of filaments and the sulfuric acid converts them into cellulose. The glucose helps to prevent crystallization of the salt formed during precipitation and also gives pliability and softness to the yarn. The zinc sulfate gives added strength to the yarn (see below) and is also responsible for its serrated cross-section, for if it is omitted, the filaments are round or oval and do not accept dyes as well. Sodium sulfate or sodium acid sulfate is always formed in sulfuric acid baths, due to the sodium hydroxide in the viscose. Its concentration is maintained within the limits given above.
The yarn after spinning is impure and relatively weak. Purification consists of washing, desulfurizing (sometimes called sulfiding), bleaching, and washing. The first wash is with water, the second is with sodium sulfide solution, the third with slightly alkaline sodium hypochlorite (followed by neutralization with dilute hydrochloric acid), and the final wash is with water again. Continuous spinning has been achieved by elimination, where possible, of the desulfurizing and bleaching steps.
The regenerated cellulose in the final product is identical to the cellulose from which it came except that the long cellulose molecules have been partly hydrolyzed and degraded into shorter, although still very long, molecules. It should be borne in mind that fibers can be made only from long molecules, and that, during and subsequent to the manufacture of a fiber, hydrolytic conditions should be avoided. Some degradation is inevitable, but means to minimize this considerably have been found. In viscose rayon, the cellulose molecules are about one third as long as those in the original cellulose; i.e., the initial degree of polymerization of about 1,100 has been reduced to one of about 350.
The tensile strength of ordinary viscose fiber varies between 25,000 and 50,000 psi; however, the tensile strength drops about 50 per cent upon soaking in water.
High-strength or high-tenacity viscose achieves its greater strength (twice that of ordinary viscose) from orientation of the molecular crystallites by cold stretching during regeneration. Complete regeneration of the coagulated filament is delayed until the filament can be stretched from 50 to 250 per cent, the lower range being used to produce medium-tenacity yarn. The zinc ions added to the bath react with the cellulose xanthate to yield a zinc cellulose xanthate which acts as an outer skin in the form of an elastic gel. This gel decreases the rate of regeneration by the sulfuric acid and facilitates stretching. The presence of zinc ion also appears to improve fatigue resistance of the yarn. For high-tenacity rayon, the concentration of zinc sulfate in the regeneration bath may run as high as 10 per cent, and it is likely that the concentration of sulfuric acid is higher also-from 45 to 75 per cent.
Viscose fibers find more than half their market in tires and other industrial uses; most of the remainder goes into apparel and household fabrics. About nine out of every ten tires made in the United States contain rayon cord; half of this tire cord is used in truck tires. High-strength viscose rayon, such as Cordura, is used in the manufacture of tire cord for heavy-duty purposes. The total amount of yarn presently produced is close to a billion pounds per year. Thus, it is not surprising that the viscose industry is the largest producer of man-made fiber and is the chief consumer of carbon disulfide and a major consumer of sulfuric acid and caustic soda. Viscose is lower in price than all other staple fibers, natural or synthetic.
Staple rayon or "fluff" is made in the same way as filament rayon except that a larger nozzle with from 2,500 to 4,000 openings is used for the spinneret. This produces a ropelike product which is cut into short lengths and then fluffed by blowing air through it. The staple is packed in bales like raw cotton and is used on standard cotton-weaving machines. About one fifth of the viscose rayon made is produced as tow or staple fiber.
In Europe, several regenerated cellutosic fibers are made which are like cuprammonium or viscose fibers but differ in after-treatment or source. Tenasco and Durafil are British products which have higher tenacities than normal rayon. Their manufacture is similar to that of American high-tenacity rayon; i.e., more zinc sulfate or more concentrated sulfuric acid in used in the regenerating bath. Fortisan, both a British and an American fiber, is made by stretching cellulose acetate in steam under pressure and then saponifying the stretched yarn to yield regenerated cellulose. It is the most highly oriented and strongest of any of the modified natural and synthetic fibers yet made, having a tensile strength of almost 140,000 psi. Since all three fibers have lower extensibilities, moisture regain, and affinity for dyes than normal rayon, they are used only when their greater strength is necessary, as in tire cords, except in the United States, where Fortisan is used in draperies and linings, presumably not only because of its strength but also because of its resistance to sunlight. In Germany, a wool-like staple fiber, known as Lanusa, is made by a combination of the viscose and cuprammonium processes, which gives it a round cross-section and a wool-like appearance and feel.
REGENERATED CELLULOSE FILM
Regenerated cellulose film, or cellophane, is the most widely used thin, transparent packaging material. It was first made in France as early as 1908. Small quantities were imported into the United States until 1924, when American production began.
The method of manufacture of regenerated cellulose film is almost identical to that of viscose rayon. The principal difference is the method of extrusion. Instead of forcing viscose through spinnerets, it is extruded from a casting machine into the acid coagulating bath.
The film produced is too brittle for commercial use without the addition of plasticizer. Suitable plasticizers or softeners improve flexibility and decrease the tendency for the film to rattle. Water is probably the most efficient plasticizer; to incorporate it into the film, the sheet is passed through a glycerol bath, after removal of sulfur and bleaching. The glycerol itself has some plasticizing effect but, being hygroscopic, also holds some water in the sheet. Approximately 7 per cent glycerol is absorbed by the film. Similar hygroscopic plasticizing agents, including ethylene glycol, glucose, and other polyhydric alcohols, are also used. The film is finally dried by passage over heated rolls.
Regenerated cellulose film is very sensitive to changes, in relative humidity and shows appreciable dimensional variation with such changes. Further, water vapor is readily transmitted by the film, and the plasticizers used are readily leached out. As a result, when the film was first used for food wrapping, it was found that baked goods, in particular, dried out too quickly. A moistureproofed sheet was developed in 1927 which opened up thousands of new uses for the product.
The usual method of moistureproofing consists in coating the sheet with a very thin layer of lacquer, which includes cellulose nitrate as the film-forming ingredient, a plasticizer for the nitrate, a resin, and a wax. The wax, usually paraffin, is responsible for most of the moisture-vapor-transmission resistance of the coating. Approximately four fifths of regenerated cellulose film is moistureproofed.
A major reason for using unsupported films is their transparency. Cellulose films are widely used to protect and improve the sales appeal of merchandise. Cellophane transmits most of the ultraviolet light as well visible light. A newer, similar packaging material (Sylphwrap) is claimed to exclude ultraviolet light; hence, this film should offer great protection against rancidity. In order to seal cellulose film to itself for packaging uses, a large amount is coated with heat-sealing lacquers which usually are also formulated to be water-vapor resistant.
Regenerated cellulose film is also produced as seamless tubing (Visking) for sausage casing. Although edible, it is not assimilated by the body.
Cellulose film is widely used in the manufacture of transparent, pressure-sensitive tape, such as Scotch tape and Texcel. The tape is usually coated with an adhesive mixture of an elastomeric polymer and various compatible resins, including rosin and coumarone resins or their hydro-genated derivatives.
Regenerated cellulose film does not have an exclusive field in the packaging market. Cellulose acetate, ethyl cellulose, polyethylene, Pliofilm, saran, and vinyl polymers and copolymers are widely used in unsupported films. Glassine and waxed papers, thin metal foils, and many laminated combinations of film, paper, and foil also compete for the very large and rapidly growing packaging business.
So far as is known, cuprammonium film has been made only in Europe, and there only on a small scale and in light film gauges. It has been produced there principally for the electrical industry and the millinery trades.
CELLULOSIC DERIVATIVES
Cellulose Esters
Cellulose Nitrate
Cellulose nitrate (often incorrectly called nitrocellulose) is the only inorganic ester of cellulose of large-scale commercial importance. Although cellulosic materials had been nitrated as early as the 1830's , Schonbein in 1845, nitrated cellulose with mixed acid and is generally credited with the founding of the industry. Twenty years later, Abel found a way to improve its stability sufficiently to allow its manufacture and storage on a commercial scale.
Cellulose nitrate was for a long time made almost exclusively from cotton and cotton linters. The great demands of wartime needs for more cellulose nitrate for propellant uses was responsible for the rapid development of a source of cellulose from wood, and by World War II, most of the cellulose nitrate made in this country (and much elsewhere) came from coniferous wood pulps.
The most important step in the manufacture of cellulose nitrate is the nitration process. The nature of the final product depends upon the degree of nitration and amount of degradation, as shown in Table 1.
Characteristics of Cellulose Nitrates
Table 1.
| Nitrogen Content % |
Nitrate Groups per Anhydroglucose Residue |
Applications |
| 10.6-11.3 |
1.8-2.0 |
Plastic molding (DP ca. 500-600) |
| 11.5-12.2 |
2.1-2.3 |
Lacquers and cements (DP ca. 150-200) |
| 12.1-13.6 |
2.3-2.9 |
Guncotton for smokeless powders (DP ca. 2,000-3,500) |
Assuming complete nitration for purposes of illustration, the following summarizes the principal reaction:
[C6H7O2 (OH)3]n + 3n HONO2 ] [C6H7O2 (ONO2)3]n + 3nH2O
This reaction differs from most cellulose reactions in that it is an equilibrium reaction, the extent of nitration at equilibrium being controlled primarily by the composition of the mixed acid. The completely nitrated ester (14.14% nitrogen) is unstable and is not manufactured commercially. It can be seen from Table 1 that commercial cellulose nitrates are made in a wide range of degrees of polymerization, which, in turn, directly controls the viscosity of solutions.
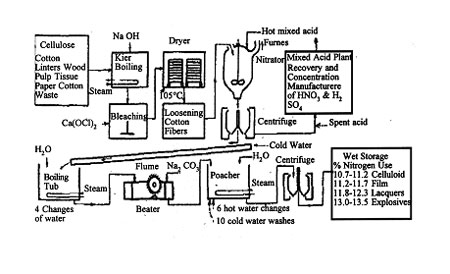
Several methods of nitration have been employed industrially, but for many years the most widely used process has been the so-called mechanical dipper process. In this method, about 1,600 pounds of mixed acid is charged to a dipper, which is a cylindrical iron or acid-resistant-steel tank fitted with two vertical, slowly rotating, opposed paddles. The mixed acid consists of about 50 to 60 per cent sulfuric acid, 20 to 25 per cent nitric acid, and 10 to 20 per cent water, variations depending upon the intended use of the ester. About 30 to 40 pounds of purified and dried cellulose are added. Larger quantities have been used, but care must be taken to control the exothermic reaction. The large excess of acids is used to minimize concentration changes during the reaction. After about 30 minutes of nitration, the whole mixture is discharged through a valve and pipe in the bottom of the dipper to a centrifuge below, where most of the spent acid is removed. The cellulose nitrate is then carefully washed and stabilized (by boiling in water for several hours), then dehydrated by displacing the water with an alcohol while compressed, into blocks in hydraulic presses. The blocks, containing about 30 per cent alcohol by weight, are shredded and the alcohol-wet nitrate is packed in steel drums for shipment.
For use in lacquers (its principal use in peacetime), the cellulose nitrate must be degraded considerably in order to obtain products of low solution viscosity. This is accomplished by digestion of the nitrate in water under heat and pressure. This was originally a batch process but has for many years been done by pumping an aqueous suspension of nitrate through a long pipe, heated at one end and cooled at the other.
Cellulose nitrate is fairly brittle and is not useful as a plastic or film-forming ingredient unless properly plasticized. For fabrication, camphor (the original plasticizer discovered by Hyatt) is still used, along with many others. The moist nitrate is kneaded with plasticizer, stabilizer (urea or amines), and pigments, if desired, and homogenized on heated rollers, which also drives off part of the alcohol. The sheets thus formed may be further dried and broken up to form molding powders or may be welded together by application of heat and pressure to form blocks from which sheets (celluloid) are sliced. These must be seasoned under controlled temperature and humidity to insure good dimensional stability.
From the sheets (Pyralin), many useful products are made, including aircraft accessories, buttons, clothes' hamper tops and coverings for toilet seats, decorative and advertising novelties, eyeglass frames, shoelace tips, table-tennis balls, pen and pencil barrels, and women's high-heel coverings.
The specific gravity of the plastic is about 1.35, unpigmented. It is tough, durable, and pliable, and can be readily formed, cemented, and machined. It has excellent dimensional stability and low water absorption. Although resistant to water, hydrocarbons, and dilute acids and alkalies, cellulose nitrate is not resistant to oxygenated solvents or strong alkalies and oxidizing agents. It is also very flammable. It is for this reason, in particular, that other cellulose esters have replaced the nitrate in many otherwise useful applications.
Pyroxylin is a term frequently used to designate products containing cellulose nitrate of soluble grade (less than 12.5 per cent nitrogen). Collodion is a viscous solution of cellulose nitrate in ether and alcohol, widely used in the past as a temporary bandage or coating for cuts.
CELLULOSE ACETATE
Cellulose acetate is the organic ester of cellulose made in largest quantity. It was first reported by Schutzenberger in 1865. At the end of the century, Cross and Bevan in England discovered milder acetylating conditions, and Miles in 1905 in the United States differentiated between the completely acetylated and the partially hydrolyzed product. Sulfuric acid was employed as a catalyst for the esterification reaction as far back as 1879 and remains the standard agent for commercial use. Cellulose triacetate was first made in the United States in 1914. Since chloroform was the only readily available solvent for the triacetate, the early production was very hazardous.
There is an important difference between the nitration and the acety-lation of cellulose. Only cellulose nitrate, of the commercially important cellulose derivatives, reaches equilibrium with the reaction components prior to complete substitution. For this reason, control of mixed acid composition used in nitrating makes it possible to vary the degree of nitration of cellulose. However, for organic esterifications which occur in nonaqueous media, it is not practicable to stop esterification short of complete reaction. This is why the cellulose acetate made commercially always starts with the formation of cellulose triacetate, often called primary cellulose acetate for this reason. Attempts for an intermediate degree of acetylation yield only mixtures of the triacetate and unreacted cellulose.
Commercially, the triacetate is hydrolyzed to a stage part way between the triacetate and the diacetate, yielding a product no longer soluble in chloroform but soluble in acetone, a more convenient, cheaper, and less toxic solvent. This is the method which has made possible the large-scale development of cellulose acetate as a plastic and a fiber, although, quite recently, the triacetate has been used in increasing amounts in molding powders, films, and fibers of high softening points.
The manufacture of cellulose acetate is almost exclusively based upon the acetylation of cellulose with acetic anhydride, using sulfuric acid as a catalyst. Zinc chloride has also, to a limited extent, been used as a catalyst. The reaction is conducted in the presence of glacial acetic acid and is not quite as simple as it may seem. The sulfuric acid reacts rapidly with the cellulose when the acetic anhydride is added at the beginning of esterification, whereas the acetylation reaction is slow. During the process, a trans-esterification reaction takes place while acetyl groups slowly replace sulfate groups. Both reactions have a preference for the primary hydroxyl group of the anhydroglucose unit. The acetic acid is present as a reaction medium for the reactants and the cellulose acetate and also serves to swell the cellulose fibers to insure complete and uniform reaction. In a variation of the process, methylene chloride is also used as a solvent.
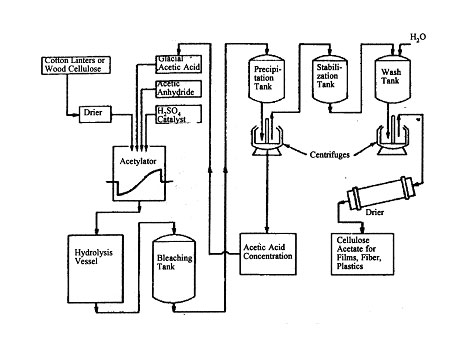
Almost all cellulose acetate is made by the solution process. Before acetylation, the cellulose is "activated" by treatment with glacial acetic acid with, often, part or all of the esterification catalyst. This appears to cause intramicellar swelling of the cellulose, so that the hydroxyl groups are made available for reaction. It is then treated with an excess of acetic acid and acetic anhydride. After mixing, the sulfuric acid is added, and the strongly exothermic reaction begins. The containers are usually cooled and the temperature kept below about 30°C in order to obtain a completely homogeneous product. The reaction is stopped by addition of sufficient water or aqueous acetic acid to make a solution of about 95 per cent acetic acid. By hydrolyzing rapidly at an elevated temperature, some chain degradation can be made to occur. If minimum degradation is desired, the hydrolysis is conducted more slowly at a lower temperature. In either case, an acetone-soluble product is obtained. After sufficient hydrolysis has taken place, the secondary acetate is slowly precipitated by addition of excess water or dilute acetic acid.
Two techniques are used to control temperature in the acetylation reaction. The older method is to conduct the acetylation in heavy-duty mixers of the Werner-Pfleiderer type. The mixers and sometimes the blades are water-cooled. The later technique utilizes a low-boiling solvent, usually methylene chloride, to absorb the heat. The boiling solvent is returned by reflux to the vessel. This is the same technique employed in the solvent method for making alkyd resins (Chap. 8), except that a much lower temperature is maintained.
A large volume of dilute acetic acid is obtained as a by-product of the process, and efficient recovery and reconcentration processes are necessary for economic reasons. Solvent extraction and azeotropic distillation methods are currently employed.
The secondary acetate varies in acetyl content, depending upon amount of hydrolysis, which, in turn, is determined by the intended use of the product. Table 2 summarizes the common types of acetate made.
Characteristics of Cellulose Acetates
Table 2.
| Acetyl Content % |
Ester Groups per Anhydroglucose Residue |
Applications |
| 36.5-38.5 |
2.2-2.3 |
Plastic molding (DP ca. 175-350) |
| 37.5-39.5 |
2.3-2.4 |
Fibers and lacquers |
| 39.5-41.5 |
2.4-2.6 |
Films and tapes |
The secondary acetate may be dissolved in acetone to form a colorless, viscous solution containing about 25 to 35 percent acetate. This is known as "dope," and it was in this form that the first large-scale use of cellulose acetate was made-as a coating for the fabric wings of aircraft in World War I so that they tightened up and became impermeable to air. After the war ended, the oversupply of acetate caused a period of intensive research to find new uses for it, and from this developed a method of converting it into an artificial silk, later known as acetate rayon, now known simply as acetate.
The dope, made as above, is passed through spinnerets into (rarely) a precipitating bath or (usually) a stream of air at about 100°C. The solvent evaporates and leaves continuous filaments of acetate which are spun under tension to increase the strength of the fiber by orientation.
Since most of the hydroxyl groups in the cellulose have now been esterified, the fiber is less hygroscopic and more water-repellent than rayon. For the same reason, the product is more susceptible to swelling or dissolution in organic solvents, however. Acetate has a greater elongation than rayon, both in the dry and wet states. Acetate yarn is unaffected by dilute solutions of weak acids, but concentrated solutions of strong acids will decompose it. Concentrated organic acids, such as acetic and formic, will cause swelling or dissolution of the fiber. Dilute solutions of alkalies have litte effect, but strong alkalies will cause saponification and transform the acetate into a regenerated cellulose form (Fortisan). It is attacked by strong oxidizing agents.
The tensile strength of acetate varies between 22,000 and 30,000 psi and drops about 40 per cent upon soaking in water.
Cellulose acetate fiber has a soft, warm texture. It is used in all kinds of women's wear, and in men's wear, including ties, hose, suits, and bathrobes, alone or in blends. It is also used in draperies and upholstery fabrics. Like most man-made fibers, it is resistant to bacteria and mildew.
Very recently, a new yarn (Arnel) has been announced which is reported to be cellulose triacetate. The method by which it is spun has not yet been disclosed. It is reported to make a light fabric which washes easily and which can be heat-treated to retain creases and pleats easily.
Cellulose acetate molding compositions (e.g., Celanese acetate, Plastacele) are prepared by mixing the precipitated material on heated compounding rolls with plasticizers, dyes, and pigments. The doughy mass after complete mixing is then rolled into sheets or slabs and blanked out into molding forms or granulated to give molding powder.
In contrast to cellulose nitrate, cellulose acetate is a light-stable, relatively nonflammable material. It is thermoplastic in nature and is suited for injection molding, whereas cellulose nitrate plastics, unstable in the presence of heat, are not. Cellulose acetate may be molded without the addition of a plasticizer; however, the unplasticized material possesses an extremely high working temperature. At very high temperatures, decomposition of the cellulose occurs. The physical and chemical properties of unplasticized acetates are not vary good; they are brittle and their mechanical strengths are low. Unlike cellulose nitrate, there is no one plasticizer that is suitable for all applications of cellulose acetate.
The useful plastics compositions contain from 60 to 85 per cent of cellulose acetate. With an increase in the amount of plasticizers used, the tensile strength and hardness decrease, and the elongation, impact resistance, and cold flow increase. Plasticizers also lower the melting point to a workable temperature and improve such properties as water and chemical resistance. Cellulose acetate plastics are resistant to dilute acids, gasoline, mineral and vegetable oils, and ethyl alcohol. Their resistance to water is not as good as that of other cellulose derivatives, a fact which has militated against applications where weathering is encountered.
Cellulose acetate flake is also utilized in the manufacture of surface coatings, sheet, rod, and tube stock, and transparent films and foils.
Lacquers made from cellulose acetate are used for insulating purposes, for sealing bottles, for coating colored light bulbs, for coating textiles, papers, and metal foils, for preparing motion picture screens, and for numerous other purposes.
This sheets of cellulose acetate are used as transparent wrapping material, and thicker strips are used as safety film, replacing the hazardous cellulose nitrate. Packaging films are made in many gauges. The film does not shrink or stretch, is waterproof, germproof, and greaseproof, and is unaffected by humidity or temperature changes. It presents an excellent surface for printing and can be cemented or laminated easily.
One other large use of acetate is that for tape for magnetic sound recording. In 1955, approximately 6 billion feet of magnetic tape were manufactured, of which about 90 per cent was made from cellulose acetate. The rest was either the cheaper, paper-backed type or the newer, more expensive, but rapidly growing polyester (Mylar) type. These tapes are usually one mil thick, are made in varying widths, and are coated on one side with a very thin layer of iron oxide in a vinyl resin binder. Tape recording is used for home amusement, radio broadcasting, motion pictures, and for diverse scientific applications. Recently, it has even been used for the magnetic recording of television pictures.
CELLULOSE PROPIONATE
Although cellulose propionate has been known for many years, com-mercial production was undertaken only recently because of the previous unavailability of propionic acid and anhydride in quantity and at a low price. It is now available under the name Forticel for use in plastic molding powders.
Its manufacture is similar to that of cellulose acetate except for suitable modifications of the process to compensate for the lower activity of propionic anhydride. Like cellulose acetate, mild acid hydrolysis yields a product of improved solubility in such solvents as acetone and dioxan. As expected, the larger fatty acid radical produces a cellulose ester of greater softness.
Cellulose propionate thus requires less plasticizer than the acetate for the same flexural strength and has a higher impact strength. It is dimensionally stable, has good surface hardness without brittleness, and is odorless, even at high temperatures. It is being evaluated and used for such applications as fountain pen barrels, telephone and appliance housings, and football helmets.
HIGHER CELLULOSE ESTERS
Cellulose butyrate can also be made by a procedure similar to that for the acetate, including the acid hydrolysis step to produce an acetone-soluble product. Due to its increased softness and lower softening point, compared to the acetate and propionate, it has not been offered on a commercial scale.
The higher esters of cellulose cannot be made in good yield by the process used to make the lower esters. Treatment of cellulose with chloro-acetic anhydride and magnesium perchlorate or with pyridine or pyridine hydrochloride as catalysts is usually necessary. These higher esters are not, at present, of commercial importance.
MIXED ESTERS OF CELLULOSE
Intensive studies have been made of the properties of mixed esters of cellulose in an effort to overcome the deficiencies of each of the straight esters. Cellulose nitrate-acetate was once manufactured but is no longer in production. Of greater interest are the mixed organic esters, especially those of the acetate, propionate, and butyrate. They are prepared by activating cellulose with glacial acetic acid, then reacting the product with mixtures of acids and anhydrides. Each of these products can vary with respect to the total degree of esterification, the ratio of the acyl radicals present, and the molecular weight.
Competitive to cellulose propionate is cellulose acetate-propionate (Tenite III, Hercose AP), developed for the manufacture of tough, dimensionally stable molded parts. The properties and applications are similar to the other cellulose esters. It is used in some surface coatings and in extruded filaments that are interwoven with cotton in the production of special fabrics for fused shirt collars.
The mixed ester made in largest quantity, cellulose acetate-butyrate, is made by esterifying cellulose with a mixture of acetic and butyric anhydrides. The acetate-butyrate most generally used is a material of about 14 per cent acetyl and 36 per cent butyryl content, having a degree of polymerization of 250 to 350. Much less plasticizer is required than with cellulose acetate.
This mixed ester combines the thermal stability of the acetate (Tenite I) with the better moisture resistance and improved dimensional stability of the butyrate. Since the acetate-butyrate retains the desirable features of moldability of the acetate and, in addition, shows great improvement in moldability around inserts, it is widely used in injection molding. It is also extensively employed in the form of extruded strips and tubes for numerous uses.
Cellulose acetate-butyrate shows better weather resistance than the acetate, partly because of lower water absorption, partly because of better compatibility and retention of resistant and nonvolatile plasticizers.
Cellulose acetate-butyrate is used in hot-melt coatings for paper and textiles; by incorporation of waxes and resins, coatings may be made with high impermeability to moisture. Both the acetate-butyrate and the acetate-propionate are used in the manufacture of safety film.
The use of strippable protective coatings has received increased attention in recent years. This type of covering is generally applied by dipping the article to be protected into a molten compostion and withdrawing it. The dipped article receives a uniform, heavy coating which quickly sets on cooling to allow immediate storage or packing for shipment. No volatile solvents are used, and so solvent and recovery are not required. The previous practice of wax or grease coating, followed by paper covering, is thus superseded by a single-step operation. Not only is the dip covering quickly applied but it is removed with equal ease. A cut is made to start the "peeling," whereupon the whole covering may be removed, generally in a single piece. This type of coating not only excludes water vapor and prevents corrosion but also protects against handling damage. Of the several thermoplastic polymers used in these coatings, ethyl cellulose and cellulose acetate-butyrate are outstanding because of their heat stability, compatibility, and high strength. One of the most promising applications of acetate-butyrate is for piping, especially in petroleum production. Corrosion problems due to salt water, sour crude, and electrolytic action have been successfully overcome with this plastic. Another advantage in the use of acetate-butyrate pipe is its lower frictional resistance to fluid flow. Flow through extruded acetate-butyrate pipe is approximately 40 per cent more than through clean steel pipe for the same head loss. The country's longest plastic pipeline-nearly ten miles of 3-inch pipe-is now in service transporting crude oil in Montana. Made of cellulose acetate-butyrate, it took only about twelve man-days to lay the entire 52,300-foot line.
CELLULOSE ETHERS
In many of its reactions, sodium or alkali cellulose behaves as an alcoholate, RONa, e.g., in its ability to form ethers upon treatment with alkyl halides. Methyl, ethyl, and benzyl celluloses have been extensively investigated and, more recently, carboxymethyl cellulose and hydroxy-ethyl cellulose have been developed. In general, the method of manufacture involves: (1) Preparation of alkali cellulose from linters and 50 to 75 per cent aqueous caustic, (2) concurrent degradation of chain length to the desired degree of polymerization, and (3) treatment at 150° to 200°C in an autoclave with the appropriate etherifying agent.
METHYL CELLULOSE
Methyl cellulose is prepared by a process similar to that for ethyl cellulose, substituting methyl chloride or sulfate for the corresponding ethyl analog (see Ethyl Cellulose). Trimethyl cellulose is not readily obtained by this method, but the partly methylated derivative has the most useful properties so that the completely methylated compound is not desired. Methyl cellulose (Methocel) is produced in several viscosity grades containing from 1.2 to 2.0 methoxy groups per anhydroglucose unit. The commercial product possesses the unsual property among hydrophilic colloids of being soluble in cold water but insoluble in hot. Upon heating, gelation occurs between 40° and 70°C, the precise temperature being a function of molecular weight and concentration.
Six to seven per cent methyl cellulose dissolved in water produces useful thickening and dispersing agents. In this form, it is used in the formulation of textile printing pastes and in cosmetic preparations and dentifrices. Alone or blended with water-miscible proteins or starches, it is used as an adhesive. Films deposited from methyl cellulose solutions have been used to coat paper. Water-resistant films can be prepared by the addition of water-soluble urea- or melamine-formaldehyde resins. As a thin film, methyl cellulose is highly resistant to greases and oils. Since it is edible, methyl cellulose has been used in salad dressings and other thickened food products.
Recently, a modified methyl cellulose has been produced, which combines many properties of the organo-soluble cellulose derivatives with the properties of the hydrophilic synthetic gums. This compound is repared by reacting alkali cellulose with both methyl chloride and propylene oxide to form an hydroxypropyl methyl cellulose. The reaction product is purified by successive washings with boiling water.
This product is nonionic, surface active, thermo-gelling in aqueous solution, and can be formed by heat fabrication techniques normally possible only with plastic-type compounds. With this mixed ether of cellulose, it is possible to extrude, mold, or hot cast from nonaqueous solvents a shaped object that is water-soluble.
ETHYL CELLULOSE
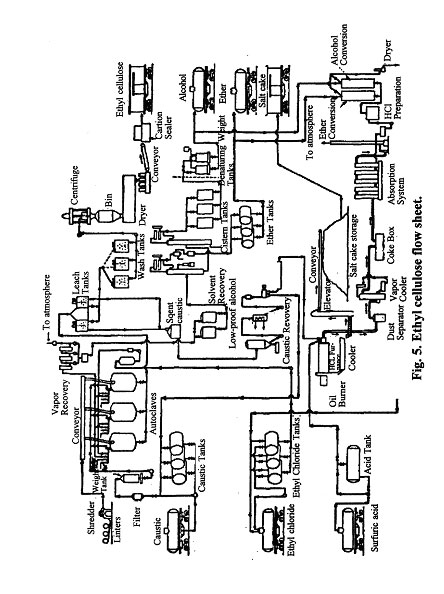
Ethyl cellulose is manufactured in greater quantity than any of the other cellulosic ethers; hence, its manufacture will be described in greater detail. The first step in the preparation of ethyl cellulose is the formation of alkali cellulose. One method consists of the following steps: After removal of the excess alkali, the wetted material is conveyed to the reactors. Ethyl chloride and excess caustic are added to the reactors. The temperature is raised to 175° to 200°C, and the reaction allowed to continue for a specified length of time. By control of temperature, pressure, reaction time, and ratios of raw materials, it is possible to control the ethoxy content and the viscosity of the product The over-all reaction for a degree of substitution of one is:
[C6H7O2 (OH)3NaOH]n + nC2H5Cl ] [C6H7O2(OH)2 (OC2H5)]n + nNaCl + nH2O
When the reaction is completed, the ether and alcohols that are formed as by-products of the action of the caustic on ethyl chloride are flashed off, the vapors scrubbed with sulfuric acid, and the solvent condensed and recovered. As the solvents are removed from the reactors, ethyl cellulose precipitates in a liquor consisting of excess caustic and salt. This slurry is blown to tubs in which the caustic is drained off and the ethyl cellulose is given a preliminary washing. The large granules of ethyl cellulose are then dropped into disintegrators where they are ground to a uniform size. This grinding makes it possible to remove additional salts and caustic by an additional wash and stabilizing treatment. Excess water is then removed in a centrifuge, the product is dried, blended, and stored according to grade. The alcohol and ether formed as by-products are recovered and reconverted to ethyl chloride.
A more recent method of preparation is as follows: The caustic solution is expelled from the alkali cellulose and all traces of free alkali removed by washing with ethyl alcohol, using 95 per cent alcohol for the first washes and absolute alcohol for the last in order to remove all traces of alkali. This method practically eliminates the formation of ethyl ether from the ethylating agent. The alcohol-wet alkali cellulose is suspended in additional alcohol in an autoclave and ethyl chloride gas is added under pressure to the stirred suspension. The temperature is slowly raised to 180° to 200°C under a pressure of 1,000 to 1,500 psi until no further consumption of ethyl chloride occurs, if complete reaction is desired. The final ethyl cellulose flake is obtained by precipitation from an acetone solution, thus assuring complete removal of any occuluded salt.
The properties of the resulting ether can be controlled by the degree of substitution, as is shown in Table 3.
Table 3.
Characteristics of Ethyl Celluloses
| Ethoxy Content % |
Ethoxy Groups per Anhydroglucose Residue |
Solubility and Applications |
| 14-24 |
0.5-1.0 |
Soluble in caustic soda |
| 24-33 |
1.0-1.5 |
Soluble in water |
| 33-45 |
1.5-2.3 |
Water solubility decreases to zero at 45 % ethoxy content |
| 45-47 |
2.3-2.4 |
Solubility in nonpolar solvents increases. Softening point decreases |
| 47-48 |
2.4-2.5 |
Compatible with waxes and resins; minimum softening temperatures; best commercial grade; tough, stable, and thermoplastic |
| 48-50 |
2.5-2.6 |
Soluble in nonpolar but not in polar solvents; softening temperature increases again |
Ethyl cellulose has a specific gravity of 1.14, lower than that of any other cellulose derivative, and is among the lightest plastics. It is produced only in water-insoluble grades. The most common products possess between 44 and 50 per cent ethoxy content. This degree of etherification corresponds to alkylation of about 2.2 to 2.5 hydroxyl groups per anhydroglucose unit, a range in which optimum solubility in both polar and non-polar solvents is obtained.
Since its introduction in the United States about 1935, ethyl cellulose (Ethocel) has found favor in both coating compositions and molded plastics because of several advantages which it possesses over other cellulosic plastics. It is unusually tough and flexible and, suitably plasticized, retains these characteristics even at temperatures as low as - 40°C. For any given flow quality and rigidity, it requires less plasticizer than either cellulose acetate or nitrate. Ethyl cellulose does not embrittle as rapidly as the cellulose esters as temperatures are lowered and its low-temperature flexibility has made it a useful material for nonstructural components in aircraft. An important characteristic of ethyl cellulose is its ability to be compounded with an amazing variety of plasticizers, gums, waxes, natural and synthetic resins, and solvents. By proper selection of these ingredients, almost any desired combination of gloss, weatherability, hardness, flexibility, and surface adhesion may be produced. It exhibits good resistance to alkalies and aliphatic hydrocarbons, fair resistance to dilute acids, but is decomposed by concentrated mineral acids and is soluble in most aromatic solvents, esters, and alcohols.
Ethyl cellulose exhibits excellent electrical resistance. In addition, films of ethyl cellulose have excellent flexibility and good water resistance, two factors which are of considerable importance in wire insulation coatings. The good adhesive properties of the material are apparent in its self-sealing properties and its application to low-pressure cloth laminates. Ethyl cellulose is also available for application as a hot-melt strip-coating for the preservation of metal parts.
One of its favorable characteristics is its stability to heat and light. In the case of cellulose nitrate lacquers, breakdown is accompanied by acid decomposition products which promote further decomposition. Since ethyl cellulose is an ether rather than an ester, heat resistance is notably superior to cellulose nitrate. Since it is compatible with cellulose nitrate in all proportions, it is utilized to augment a variety of pyroxylin coatings.
Ethyl cellulose, when compounded, may be used in countless ways. Its applications include packaging papers, wrapping foil, artificial leather, lacquers, adhesives, furniture trim, automotive parts, wire insulation, toilet articles, novelties, fabric coatings, and flexible rubberlike tubing.
Ethyl cellulose film is made by casting from an alcohol-toluene solution onto a moving, continuous surface. The film is characterized by resistance to attack by alkali and dilute acids, resistance to light discoloration, and good flexibility and toughness at low temperatures. It has relatively low water absorption and better toughness than cellulose acetate; however, since ethyl cellulose is not inherently moisture-vapor-proof, this property is added by incorporating in the formulation a moisture-vapor-resistant material, such as paraffin wax.
Ethyl cellulose has never been used on a commercial scale to produce a fiber. Its solubility in a wide variety of solvents, some of them well-known dry-cleaning agents, and its comparatively low softening point have restricted its use in the textile industry.
Benzyl cellulose is manufactured in Europe but not in the United States. Ethyl hydroxyethyl cellulose is made here and is similar in properties and applications to ethyl cellulose, except for better solubility in aliphatic hydrocarbons.
CARBOXYMETHYL CELLULOSE
Carboxymethyl cellulose was first developed in Germany toward the end of World War I, but it was not until late in the 1930's, and particularly during and since World War II, that large-scale uses began to emerge and commercial production appeared justified.
Carboxymethyl cellulose is an ether of cellulose and hydroxyacetic (glycolic) acid. The cellulose is treated with alkali, then reacted with sodium chloroacetate to yield sodium carboxymethyl cellulose. An anhydroglucose unit with a degree of substitution of one would be:
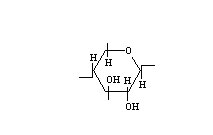
Theoretically, complete reaction would mean the introduction of three carboxymethyl groups per anhydroglucose unit. In any actual molecule, the substituent groups are scattered randomly along the chain, some on primary and some on secondary hydroxyl groups (with no units substituted in both secondary positions). The optimum combination of physical properties of the commercially available sodium salt is achieved with a substitution of 0.4 to 1.3 (0.7 being the most common). The free acid formed at a degree of substitution of 1.3 or below is insoluble in water, whereas the sodium salt at degree of substitution of 0.3 or above is soluble. Thus, upon acidification of aqueous solutions of sodium carboxymethyl cellulose, the free-acid form precipitates. The acid form is infusible and cannot be dissolved without being neutralized; therefore, the product is not widely useful. On the other hand, a stable, colloidal dispersion of the free-acid form in water can be prepared by ion exchange between an aqueous solution of sodium carboxymethyl cellulose and a strong, acid-type cation exchange resin. In this form, the free acid has unique properties which indicate its possible usefulness as a film former.
The two best-known processes for the manufacture of sodium carboxymethyl cellulose are the German batch process and the continuous process. In the batch process, the principal steps are the reaction between bleached sulfite pulp and caustic soda to form alkali cellulose, and reaction of the latter intermediate with dry sodium chloroacetate to form sodium carboxymethyl cellulose and sodium chloride. Excess caustic is neutralized with sodium bicarbonate, and the product (CMC, cellulose gum) is ground and packaged.
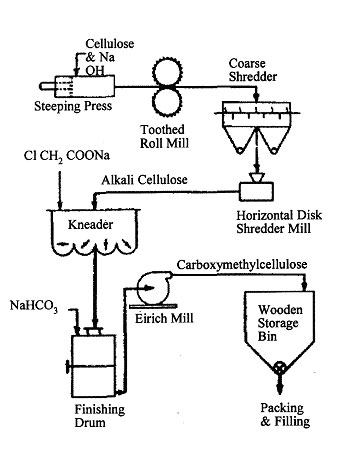
The continuous process consists of spraying powdered cellulose first with caustic solution and then with chloroacetic acid as the solid cellulose is tumbled in a rotary drum. For most industrial uses, a dried form, containing 5 to 6 per cent moisture, is produced. The wet sodium carboxymethyl cellulose from the reactor, after aging for eight to ten hours, may be dried by several methods. Flash drying yields the product in its most desirable physical form (Carbose).
Pure sodium carboxymethyl cellulose is a white powder, soluble or dispersible in water or alkaline solutions. Aqueous solutions are highly viscous, and for this reason are useful for their thickening, suspending, and stabilizing properties.
In pure form, the powdered product is odorless, tasteless, and physiologically inert. Considerable use is expected for purified grades which can be added to food, cosmetic, and pharmaceutical preparations. Carboxymethyl cellulose is an excellent stabilizer for ice cream, since, by inhibiting the formation of ice crystals, it decreases the amount of beating required for the incorporation of minute air bubbles which improve the consistency and decrease the density of packaged ice cream.
It has proved of value in water-soluble adhesive compositions, in stabilization of latices, and in ceramics, where it serves as a dispersing agent to help keep ceramic glazes in suspension. In the paper industry, it has been used to increase the bursting strength of liners, to increase the gloss on coated box boards, and to help suspend ink for subsequent removal in de-inking systems. The European textile industry has employed carboxymethyl cellulose for warp sizing; this application has not yet been very successful in the United States. Less than 1 per cent carboxymethyl cellulose added to oil-well drilling mud improves mud viscosity control and helps to plug porous formations in the bore, preventing loss of water from the mud.
Probably the material's greatest potential lies in the laundry field, where detergent improvement uses larger quantities of carboxymethyl cellulose each year. In addition, the product has been found effective as a laundry sizing. Although evaluation has not been extensive, there are indications that carboxymethyl cellulose may improve the physical characteristics of soil in somewhat the same manner as the acrylic-type soil conditioners do.
HYDROXYETHYL CELLULOSE
Although hydroxyethyl cellulose has been described in the technical literature for the past thirty years or more, few uses in commerce have developed. Depending upon the degree of substitution, three general types have been made-one of low substitution, dispersible in chilled caustic solutions; a medium degree of substitution, soluble in caustic at room or elevated temperatures; and a water-soluble grade, of a higher degree of substitution.
Hydroxyethyl cellulose (Cellosize) is made by metathesis between ethylene chlorohydrin and alkali cellulose or by direct reaction of ethylene oxide upon cellulose itself. The over-all reaction for a completely substituted product can be written as follows:
[C6H7O2(OH)3]n + C2H4O ] NaOH aq. ] [C6H7O2(OC2H4OH3]n
The function of the sodium hydroxide is to swell the cellulose fibers uniformly and to catalyze the etherification.
The product varies in appearance and properties from a fibrous, water-insoluble material, superficially similar to cellulose, to a waxy, water-soluble material with increasing ethylene oxide content.
Hydroxyethyl celluloses have been used as filaments, in sheet form, in textile finishes and sizes, in paper coatings, as a thickener for textile printing, as fixing agents for pigments, as leather coatings, as fillers for greases, and as additions to hydraulic cement used in lining deep wells to retard its solidification at high pressures and temperatures.
A combined ether, carboxymethyl hydroxyethyl cellulose, is also, made which is similar in properties and applications to carboxymethyl cellulose, but is more compatible with various salt solutions.
CYANOETHYL CELLULOSE
By treating cellulose first with caustic, then with acrylonirile, a series of cyanoethylated celluloses are obtained. Depending upon the temperature, mole ratios of caustic and acrylonitrile to cellulose, and time of reaction, various degrees of substitution are obtained. As with products just previously discussed, cyanoethylated cellulose is an ether:
[C6H7O2(OH)3]n + nCH2 : CHCN ] [C6H7O2(OH)2 (OCH2CH2CN)]n
Alkaline hydrolysis of the product yields carboxyethyl cellulose. Depending upon the reaction conditions, which control the degree of substitution, the products may be alkali-soluble, water-soluble or organic-solvent-soluble.
A considerable amount of research has been done upon the cyanoethylation reaction, with both wood and cotton, in an effort to upgrade the properties of cellulose. Water-soluble yarns, twistless yarns, and related products have been claimed. Further, by achieving a degree of substitution of from 0.3 to 2.0 cyanoethyl groups per glucose residue (organic-solvent-soluble), it is claimed that the properties, especially the tensile strength, wear resistance, stiffness, and moisture and mildew resistance, of cotton fabrics are improved. Applications under investigation at present include the use of cyanoethylated cotton in tarpaulins, filter fabrics, awnings, fish nets, tobacco shade cloths, and similar items where permanent resistance to rot, mildew, and bacteria and improved abrasion and stretch resistance are desired.
Even paper has been cyanoethylated. The treatment improves the water resistance and dimensional stability. These properties are parti-culariy important in punched cards used in automatic business machines. The chief drawback to the cyanoethylation of cellulose in all forms so far has been its relatively high cost. It is likely that increased interest in the application of this process to cellulose will occur as the cost of acrylonitrile decreases.
Fabricating and Processing
In this chapter there will be discussed and illustrated many of the ingenious techniques which engineers have developed to transform powders, granules, sheets, and liquids into the myriad shapes and forms useful for countless applications.
Monomers and polymers (and, for that matter, all other structural materials) are of little or no practical use until the raw product from the manufacturing process has been transformed by more or less standardized fabrication and processing techniques into useful forms. Hence, the task is to convert liquids (in the case of monomers) or solids (in the case of polymers) into physical states and shapes which will be useful for the applications for which they are intended. Note here the distinction between fabrication and application. The former involves a manufacturing technique to put the raw material in the physical shape or state suitable for use. The latter term involves the use of the product for the purpose for which it was fabricated or processed.
Most attempts at classifying the diverse methods of fabrication and processing have met with small success, in large part due to the fact that many final shapes may be obtained by more than one method. A further difficulty in discussing these methods arises from the inexact and often ambiguous nomenclature which has evolved in industry. The following system of classification of fabrication and processing techniques is possibly the most satisfactory method and is based upon the physical state of the raw product, since this will, to a large extent, determine the method of processing.
Table 1.
Processing of Polymers
| Solids |
Liquids and Melts |
| Molding |
Coating |
| Extrusion |
Expanding or Foaming |
| Calendering |
Casting |
| Sheet forming |
Spinning |
| Solids and Liquids |
|
| Laminating and impregnating |
|
MOLDING
Molding is usually defined as "the process of forming in or into a particular shape." This definition is a very broad one, and the term may justly be extended to include extrusion, forming, impregnating, expanding, casting, and spinning, all of which involve a shaping operation of some kind. In order to differentiate between the types of shaping mentioned above and those involving the use of a closed mold, molding, as used in this section, will be defined as the shaping of an article from a plastic material by the application of pressure (and, usually, heat in a closed chamber.
Cold Compression Molding
Cold molding is one of the oldest methods of shaping plastic materials. The process possesses an economic advantage over most other molding techniques and for this reason has continued to be used. On a percentage basis, its use is decreasing because of the strong competition from the faster, and. in many cases, higher quality molding processes which have subsequently appeared.
This type of molded product first came into use in the United States about 1908-09, at approximately the same time as the advent of the phenolic hot-molded compounds. Both were the result of a search for a heat-resistant, molded, insulating material for use in the electrical and automotive industries. Its predecessor, porcelain, although widely used because of its fireproof, heatproof, and waterproof properties, was so brittle that it frequently could not be used in spite of its excellent insulating qualities. Other products investigated were vulcanized fiber, hard-rubber compounds, and shellac, but, due to individual limitations, all were eventually superseded by compositions made by the cold-molding process.
Cold-molding compounds may be divided into two types, depending upon their compositions and temperature limitations: nonrefractory (organic) and refractory (inorganic). In general, the binders suitable for nonrefractory cold-molding compounds are of two classes, bituminous and resinous. The bituminous class includes asphalt, gilsonite, and pitches, which are blended with drying oils. Resinous binders usually consist of phenolic resins dissolved or dispersed in solvents. This type generally has better electrical and mechanical properties than the bituminous type. Fillers used consist principally of asbestos fibers, silica, and magnesia.
(Refractory cold-molding compounds usually contain Portland cement. clay, lime, silica, or phosphoric acid as the binder, all filled with asbestos; all but the last-named binder are moistened with water before molding.)
Cold-molding compositions differ from all others in that they are manufactured at the place of molding. This is so because their preparation requires only simple mixing of readily available components and the mixed batches have a tendency to dry out or cure if not molded soon after mixing.

The molding operation is similar to hot compression molding except that the molds require neither heating nor cooling. Whenever possible, the cold-molding powder is preshapcd to the approximate shape of the finished article. The charge placed in the mold must be accurately measured, since neither flow nor flash of the material occurs during the molding operation. Since cold molding is merely a compression of the molding mixture, the change in the mold is purely physical. The pressing operation operates under pressures of from 2,000 to as high as 30,000 psi and is continued only long enough to mold the material into the desired shape. After removal from the mold, the article is transferred to an oven and is baked at temperatures of 80° to 260°C, depending upon the binder used.
The molding operation itself is very rapid, since there is no heating or cooling cycle. The low cost of the molding material and the high production rate, which usually eliminates the need for expensive multiple-cavity molds, have served to keep the cold-molding process in use.
Cold-molded parts usually lack the surface smoothness and gloss associated with hot-molded or cast plastics; even the high pressures required to compensate for the lack of plastic flow of these materials are not sufficient to reproduce accurately fine mold design, and, therefore, cold-molded articles are usually limited to simple shapes. Since the articles are primarily of value for their good electrical characteristics, appearance is not usually of great importance. Common electrical products include such items as connector plugs, switch bases, outlet covers, attachment plugs and caps, and sockets. Other items, such as buttons, checkers, and storage battery cases, are often cold molded.
Hot Compression Molding
Hot compression molding, because it is much more commonly employed than cold compression molding, is usually called simply compression molding. Its distinguishing feature is the application of pressure to a molding powder in a heated and closed mold. This fundamental idea of shaping a plastic body by heat and pressure was first applied by Baekeland to phenol-formaldehyde resins about 1907. Since that time, the art of compression molding has been continually improved and refined. Although the principle of heating and shaping by pressure in the same cavity is still actively followed, newer developments, such as transfer and injection molding, have demonstrated that the principle of heating and shaping in separate chambers is also of great value.
Thermoplastics can be molded by this technique, but then the molds must be cooled before removal of the shaped article and must be reheated before receiving the next charge. This expense, the time lost, and the development of faster and more satisfactory methods for molding thermoplastics have restricted the process mainly to the molding of thermosetting compounds. Compression molding is used, however, with some rigid and unplasticized thermoplastics which are so viscous or unstable at their maximum molding temperatures that unsatisfactory results by other techniques occur.
In the general process of compression molding, the charge is placed in a heated mold, and the mold is closed, generally under low pressure, until pressure is exerted on the material. The material becomes plastic and, as the mold continues to close, the softened material is forced to fill all the cavities of the mold. High pressure is applied to the mold just before it is completely closed in order to insure complete filling of the mold. The molded article is kept under pressure until it is cured, after which the mold is opened and the molded part is removed. Often the mold is allowed to breathe (i.e., is opened momentarily after first closing it) in order to allow gases present (air, moisture, etc.) to escape before the final closing is performed. From this brief description, it may appear that the process is a very simple one. It is true that the principle is simple, but the actual techniques used, materials of mold construction, and mold design have evolved after many years of engineering evolution. Some of these techniques and considerations will now be briefly discussed.
The mold charge may be used as supplied (i.e., loose granules, beads, chips, scraps of resin-impregnated cord or fabric), or it may be tableted or preformed. Tableting involves the pressing of the compounded resin into a cylindrical, rectangular, annular, or other simple shape having some common use, singly or in multiple, in various molds in the molding plant. In preforming, the material is roughly pressed into the shape of the molding to be produced. In rarer cases, where flow is poor (e.g., polytetrafluoro-ethylene), complete shaping of the preforms to the mold cavity is advantageous. Tablets and preforms are usually much less dense than is the final molding, but are appreciably more compacted than the original material.
Although it requires a separate operation, tableting or preforming offers several advantages. The charge may be handled with greater facility, both in loading of the mold and in preheating of the charge. A big advantage is that automatic premeasurement of the charge is accomplished. Tablets and preforms are pressed to the desired shape without heat at pressures ranging from 5,000 to 20,000 psi (cf. cold molding).
Preheating of the molding material before it is placed in the mold is a common operation. By its use, less pressure is required for molding, and the cure time is decreased (especially for thick sections); in fact, preheating is a prerequisite for plunger molding (see Transfer Molding). Further, because of the greater fluidity of the charge, preheating makes possible the molding of pieces containing intricate or delicate inserts. The heat also insures using a dry material, resulting in optimum electrical properties and reduced shrinkage.
There are three general types of preheating: high frequency, steam, and air. The first type is also known as heatronic molding and electronic heating. This process consists in preheating a plastic by placing it in a high frequency (2 to 70 megacycles per second) electric field prior to the molding operation. In such a rapidly reversing field, a tremendous amount of internal molecular friction occurs, with the resulting formation of heat. This method of heating differs from all other types in that the heat is generated within the material being heated, requiring no conduction into the material. Consequently, the inside of the plastic is hotter than the outer surface because of conduction from the surface to air, whereas the reverse is true using other methods of heating.
High-frequency preheating is in wide use because it enables a molding compound to be heated very rapidly and uniformly. As a result, more uniform plasticization and faster cures are possible than with other methods. However, the initial cost of the electronic equipment is higher.
This technique, with suitable modification, is applicable not only to compression molding but also to transfer and injection molding, to the curing of laminates and plywood, and to thermoplastic welding operations. It is employed principally for the preheating of preforms used in compression molding, thereby enabling the rate of this process to approach that of injection molding of thermoplastic materials. The lower frequencies are usually employed for the slow curing of laminates, while the higher frequencies are used for molding, where more rapid heating rates are desirable.
Steam or moist air preheating is not popular at present because it is much slower than high-frequency preheating. However, its cost of installation is less and it can be used to control material moisture content, which, to some extent, influences the shrinkage and dimensional stability of molded pieces. This is of value in molding instrument parts where great accuracy is required.
Dry-air preheating is useful when electrical properties of the product are particularly important because it dries the molding compound more effectively than all other methods. It is the slowest method of preheating and, like moist-air preheating, may cause some procuring because of nonuniform heating caused by conduction from the surface.
Molding is performed under varying conditions of temperature, pressure, and cycle, depending upon the characteristics of the molding compound used. The temperature of the charge may vary from room temperature (no preheating) to about 150°C. At higher temperatures, the rate of cure becomes too great for practical handling of the charge. Mold temperatures vary from 145° to 200°C but are generally in the range of 160° to 185°C, except for the fluorinated plastics for which the temperature may be as high as 380°C. The mold is heated by a variety of methods, which include steam, gas, electrical, and induction heating, and superheating of water. The optimum temperature of molding differs for the various molding compounds and is fairly critical.
The control of pressure is equally important. The pressure exerted during molding is based upon a knowledge of the cross-sectional area of the molded pieces and, in molds where "flash" occurs, from the total area of molding material upon which pressure is exerted. Usually, a combined low-pressure and high-pressure system is used in order to supply a low pressure of 300 to 500 psi during closing of the press and average pressures of 1,500 to 8,000 psi during molding. These pressures are adequate for most materials and, at the same time, prevent fatigue of the steel members of the mold, which can break down the case-hardened metal and cause dimensional inaccuracies in the molded product. Pressures for preforming, on the other hand, may run as high as 40,000 psi, since this operation is performed at room temperature and the molding material has no plasticity.
Molding cycle times depend upon the shape and thickness of the molded article, the type of molding material, and the technique used; they usually vary from a few seconds to several minutes.

Molding may be performed in hand-operated, semi-automatic, or fully automatic presses, depending upon the rate desired and the nature and number of articles to be made. Hand molds are used only for intricate pieces and for very short runs. The assembly, charging, disassembly, and discharging are done by hand. In semi-automatic molding, the mold is permanently mounted in the press and the molded pieces are ejected automatically. This type of molding is most popular, particularly for large pieces. In fully automatic molding, the operator merely supplies material for each machine and periodically removes accumulated pieces; the loading, molding, and ejection steps are automatic. This method is used for extremely long runs of small pieces, such as closures and tube bases, and, in such cases, is economical in spite of the initial high cost of the equipment.
Compression molds are generally of four different types of design. A fully positive mold is one which fully confines the material, the travel of the force being limited only by the material in the mold, since the mold has no land or cutoff. A very small amount of material may escape between the force and the die. The mold charge must be very accurately measured, since the amount determines the thickness of the piece. This type of mold is used when maximum density in a product, such as the high-impact plastics, is desired, since the total molding pressure is applied only to the projected area of the molded piece. This type of mold is used very rarely with thermoplastic materials. It is also not used for multiple-cavity moldings, because the dimensions of the molded articles vary with the charge. Since it usually is impractical to measure mold charges exactly or to confine them closely, there is always some necessary expulsion of surplus or even undesired loss of charge from the mold cavity. This excess is called flash. The quantity of charge must be sufficient to produce a molding of proper density under proper pressure, yet allow for flash.

Thus, a flash-type mold is one in which the material is not confined within the mold cavity until the instant of final closing. A relief must be provided around the cavity on either the upper or lower part of the mold or sometimes on both; thus, only a comparatively small area, called the cutoff, is in contact when the mold is completely closed. Once the mold is completely closed, however, the cutoff prevents positive pressure from being exerted on the contents. As a result, the density of the piece is less. The overcharge, usually about 3 per cent, escapes through the narrowing space between the lands as the mold closes. This type of mold is less expensive and forms pieces of constant dimensions. It is generally used to produce thin, flat pieces.
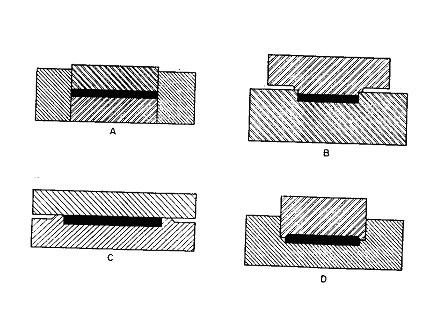
Combination of the positive and flash-type molds are used, the commonest being usually referred to as a semipositive mold. This is the type of compression mold most frequently used. As with the fully positive mold, the upper and lower parts telescope; like the flash type, however, the material is partially confined and has some provision for overflow of the slight excess of charge. The mold produces a piece of maximum density, in as much as the material is under positive pressure during the last portion of mold travel and just before the mold has seated on the cutoff edge. Further, positive pressure is maintained without the necessity for such accurate weighing as is desirable or necessary in the positive type of mold.
A landed positive mold, which may be either externally or internally landed, is generally used where it is necessary to form a radius, bevel, bead, or some other projection. This type of mold, like the semipositive mold, is a combination of the fully positive and flash types, and differs from the semipositive in that only a small horizontal flash is obtained, full pressure being maintained on this flash and the charge. As with the other types of positive molds, the halves of the mold telescope to some degree, thus providing better transmission of pressure to the material than in the flash type. A variation of the landed mold is the subcavity mold, which is one having more than one subcavity within the main cavity but having only one force.
Molds are generally made from steel, the quality of the steel ranging from soft mild through carbon steel to the special hard, stainless, alloyed types. Nonferrous casting alloys, such as beryllium-copper, are often employed for molding thermoplastics where intricate cavities are required and rapid wear is not encountered. Often tool steel is used for molds. It is machined, then case hardened, after which it is ground and polished. Stainless steels are used when staining of molds must be avoided, as for pastel shades of urea resins. Molds are often chromium plated, since chromium is very resistant to abrasion and corrosion and increases greatly the working life of a mold. The surface of chromium plated molds improves release of molded items, resists staining, and imparts a very smooth surface to the products.
For the production of large quantities of identical moldings, multiple molds may be used. It is usually much simpler and cheaper to use a single mold with multiple cavities, however. Electroforming over a master mold, made of metal, wood, plaster, etc., is used for both processes and reproduces the original mold form faithfully. One of the best methods of making multiple cavities, of identical shape and size, is by hobbing. This process was conceived about 1876, shortly after the Hyatts developed celluloid. Large-scale production of such items as toothbrush handles and harness buckles could be produced only in steel multiple-cavity molds. However, production of such molds by machining was not only very expensive because of the handwork involved, but it was also practically impossible to make multiple cavities of uniform size and contour by hand methods.
The hobbing method forces a highly polished and hardened steel master (called a hob) into a piece of cold, soft steel by means of a hydraulic press; the cavity formed has a surface which is as highly polished as the hob. In addition, the soft steel is so compressed that a finer grain size in the surface of the hobbed cavity results. All of the cavities produced from a master hob are of the same size and have uniformly polished surfaces.
Very high stresses are developed in both the soft steel of the blank and the tool steel of the hob, since the pressures required for hobbing ordinarily range from 50 to 200 tons per square inch of die impression. From one to six annealings of the mold are required to relieve these stresses.
It must be remembered that the dimensions of a molded piece will not necessarily be the same as the dimensions of the mold in which it was made, particularly in the case of thermosetting compounds which cure during the molding cycle. Consequently, the mold design must allow for shrinkage of the plastic during the curing operation. Further, molds should be designed so that parts can be readily and rapidly produced; i.e., the molds should be easy to load, easy to clean, and, particularly, the moldings should be easy to remove. The latter step is facilitated by providing sufficient draft or taper on the sides of the mold cavities and the plunger.
Compression molding is particularly useful for the production of parts of large area and deep draw, having relatively simple shapes. Furniture drawers, radio and television cabinets, instrument and business machine housings, and similar large objects are typical examples. This method of molding is also commonly used for the large-scale production of smaller, simple pieces, such as closures and buttons. Resins most adaptable to compression molding are the thermosetting types, such as phenoplasts, aminoplasts, and reinforced polyester resins.
Transfer Molding
One big disadvantage of compression molding is the difficulty in completely curing thick webs due to the poor heat transmission of plastics. This difficulty was overcome by the development of transfer molding, a technique developed in 1926 for molding thermosetting compounds in which use is made of a preheating chamber (transfer pot) from which the preheated molding compound is forced into the mold proper when the desired plasticity is attained.
Transfer methods may be subdivided into pot-type transfer and plunger or auxiliary ram transfer. The latter method has largely, but not entirely, displaced its predecessor, pot-transfer molding.
The plunger method uses a small diameter transfer pot in which one or more transfer plungers force the plastic into the mold. It differs from the pot-type system in that the molding material is forced through a very small runner and gate. As a result of friction generated by passage through the restricted openings, the material receives additional heating, and moldings are cured much more rapidly.
In conjunction with high-frequency preheating, plunger molding effects considerable savings by the large reduction in the length of the molding cycle. This method is the best that has been developed for molding thermosetting materials in times approaching those of the injection molding of thermoplastics; in fact, it has sometimes been called the injection molding of thermosetting materials. However, it differs from injection molding (q.v.) in that the mold is maintained at the molding temperature, since the parts, being thermoset, can be ejected from the mold without cooling. Another difference is due to the nature of the plastic-throughout a transfer process, the plastic is slowly hardening and only one shot or cycle can be completed at a time. There is some similarity between the two processes, however, since in both the plastic powder is hot and semi-plastic, and flows under pressure evenly into the mold cavity without unbalanced pressures being present to distort any inserts present. This is another advantage over compression molding.
The molding process consists in placing the molding compound, usually in pellet form, in the preheating cavity, allowing it to become semifluid, then forcing it through an orifice by means of a ram into the mold cavity. The shape of the orifice, which is tapered slightly, is critical, since the material in the orifice (the stalk) is the only flash occurring in transfer molding and it is desirable that it be reduced to a minimum. Since the stalk is very thin, it will harden first, and it thus forms a bung to the mold, so that, as soon as the stalk is hard, the ram may be withdrawn and the heating continued until the piece is fully cured. After ejection of the cured molding, the small flash stalk is broken off. Thus, an added advantage of the transfer-molding technique is the small amount of flash produced, which reduces greatly the expensive finishing operation necessary in compression molding.
Although a transfer pot provides a more effective and positive means of applying pressure to the charge, additional pressure is required to overcome resistance in the sprues, runners, and gates (the small passageways or openings through which the charge passes into the actual cavity-see Injection Molding). Therefore, compression pressures are appreciably greater than those used for compression molding-up to three times as great.
Transfer molds must be vented to allow escape of air trapped in the mold during injection of the material. The vents are small openings usually located on the same half of the mold as the runners and are located at the points which fill last, in order to facilitate the expulsion of air and other gases.
The proper amount of heat and pressure used in molding is extremely important, since the resins used are thermosetting. Although insufficient heat and pressure will cause insufficient flow, too much may cause the material to harden too soon and may possibly jam the machine. The material requirements for transfer molding differ to some extent from those for ordinary compression molding. For this reason and because a longer flow path is involved than in compression molding, materials used must cure somewhat more slowly in transfer molding. Nevertheless, when used with high-frequency preheating, transfer molding is quite rapid and a good production rate is possible.

The plastics most widely used in transfer molding are phenol-formaldehyde and melamine-formaldehyde molding compounds. Urea-formaldehyde resins can be transfer molded but, because of their greater reactivity and critical behavior in high-frequency preheating, they are not as widely molded by this process as the phenolic and melamine resins The more recent polyester molding materials are satisfactory for small articles, but their very rapid curing rate has prevented their use in larger molds and for articles where considerable plastic flow is required. Highly viscous thermoplastics, such as rigid poly(vinyl chloride), may also be transer molded.
The principal advantage of transfer molding, as has been mentioned, is that complicated shapes, using split molds and complex inserts, can be made. Other advantages of transfer molding are the extreme accuracy in location of inserts, closer tolerances possible, case of molding fabric-filled compositions, reduction of molding cycle times, elimination or substantial reduction of rejects for certain types of pieces, lower mold costs, and substantially lower finishing costs, although the loss of the cured sprue and residue in the transfer chamber may result in greater material costs than in compression operations. Also, shrinkage may be only one fourth as great as in compression molding, probably because of the higher pressures and the fact that the material has been partly cured by the time it fills the mold.
