Polyesters
PRODUCING EXPANDED AND CURED POLYESTER RESIN
Highlights of the Technological Achievement: Polyester resin is expanded and cured using monosubstituted sulfonyl hydrazide blowing agent, organic peroxide curative and certain metal promoters.
Background: It has been found that polyester resins (PER) can be expanded and cured simultaneously when employing monosubstituted sulfonyl hydrazides (MRSH), a primary organic metal salt promoter, with or without a secondary organic metal salt promoter, a surfactant and an organic peroxide or hydroperoxide at low concentrations of peroxide and promoters.
Process Conditions: This process provides low density foams prepared from liquid ethylenically unsaturated polyester resins by blending various ingredients as outlined on the listing below.
| Ingredient |
General |
Preferred |
Most Preferred |
| Polyester resin, parts |
100 |
100 |
100 |
| MRSH blowing agent, mm |
2.7-32 |
5.4-27 |
11-22 |
| Primary promoter, parts |
0.001-2 |
0.001-1.5 |
0.01-1 |
| Secondary promoter, parts |
0-0.4 |
0-0.25 |
0.01-0.2 |
| Surface active agent, parts |
0-2 |
0.5-1 .5 |
0.75-1.25 |
| Filler, parts |
0-250 |
0-150 |
0-100 |
| Organic peroxide (or hydro-peroxide) curative, [per 100 parts of (a) plus (f)] |
0.2-2.5 |
0.5-2 |
1-2 |
The following guidelines may be useful for the practitioner: if no (d) is present, then the amount of (c) is preferably at least 0.025 part. Also, if no (d) is present and the level of (c) is at least 0.01 part, then the concentration of (b) should preferably be at least 11 mm, and that of (g) greater than two parts. It should also be noted that if (b) is present at a 5.5 mm or higher level, and (g) is present at one part or more, the concentration of (c) should preferably be at least about 0.5 part in the absence of (d).
Using the proper concentrations of components, foamed polyester structures may be obtained exhibiting most preferably at least 30% or more of density reduction when compared to the nonfoamed polyester resin.
The liquid unsaturated polyester resins in the composition comprise a linear or only slightly branched polyester resin and an ethylenically unsaturated monomeric compound.
Examples of saturated polybasic acid include: isophthalic acid, orthophthalic acid, terephthalic acid, adipic acid, succinic acid and azelaic acid.
Flexible resins employ greater amounts of adipates or azeleates, while more rigid resins use phthalates, with a variety of different glycols. This process is useful for making rigid and semirigid polyester foams suitable as structural-type foams. Such resins have a formulation, for example, of about 3 to 5 mols of glycol, 1.5 to 3.0 mols of adipic acid, and 0 to 1.5 mols of phthalic anhydride, with from 1.0 to 2.5 mols of styrene or vinyl toluene.
The chemical blowing agents suitable for preparing the foamed and cured polyester are monosubstituted sulfonyl hydrazide having the structural formula RSO2NHNH2, wherein R is a hydrocarbyl radical selected from C1-2 alkyl, C5-6 cycloalkyl, C7-10 aralkyl, phenyl, naphthyl, also phenyl substituted with halogen, C1-2 alkyl, or C1-12 alkoxy.
FOAMED UNSATURATED POLYESTER RESINS WITH GEL COAT
Highlights of the Technological Achievement: A process for the production of foamed products from an unsaturated polyester resin composition, the composition comprising polyester resin, unsaturated monomer, a promotor, initiator, physical blowing agent, and compatible surfactant whereby a known gel time "T" results, foaming the composition by exposing it to microwave radiation prior to 1 T, thereafter permitting the foam structure to exotherm to obtain a fully cured product.
Background: Microwave energy has been suggested for the curing of foam materials in the past. One example of this is Jacobs, U.S. Patent 3,216,849.
While unsaturated polyester resin is mentioned, a thorough consideration of this patent will show that the system disclosed is only applicable to the polyurethane foam which is specifically described.
The Jacobs process is distinctly different from this process. With the polyester system, quick energy addition is necessary to volatilize the foaming agent and to control the rate of gelling. Stated another way, Jacobs reaction causes the foaming and the microwave source is an ancillary finishing step, while in this process the microwave energy causes the foaming and the material has no cellular structure prior to such exposure.
Process Conditions: The time of exposure to the microwave energy is sufficient to volatilize the foaming agent and to accelerate crosslinking of the resin to or close to the gel condition. Preferably the initial exposure is made between 0.2 and 0.7 T.
The process also resides in products produced according to this method. Unfilled products can be made having a density of usually 8 to 15 pcf while filled products usually have a density of 10 to 50 pcf.
In a further aspect for the production of foam products from an unsaturated polyester resin composition, the composition comprising a polyester resin, styrene, promoter, and surfactant and having upon subsequent addition of an initiator a gel time of "T," the process comprising adding initiator and a physical blowing agent to the composition, exposing the composition to microwave radiation at a time greater than 0.2 T (preferably between 0.2 and 0.7 T) and less than 1 T whereby foaming takes place prior to gellation, thereafter permitting the foam structure to exotherm to obtain a fully cured product.
From the above, it can be seen that the basic concept of the process can be simply stated. The microwave energy, when applied at the proper time in the reaction process, supplies uniform and intense heat to the resin system, thus allowing vaporization of the dissolved physical foaming agent. At constant microwave energy input, application of the energy too early in the process will lead to gross bubble formation and/or cell collapse prior to gellation. Application too late in the process will lead to premature gellation and an unworkable foam. Similarly, at a fixed point in the reaction process, exposure for too short a time will lead to low foaming levels and long gellation times outside the microwave unit, yielding bubble coalescence and cell collapse. If the exposure time is too long, excess foaming and gellation occur inside the microwave unit, yielding overheating, styrene vaporization and catastrophic cell wall rupture.
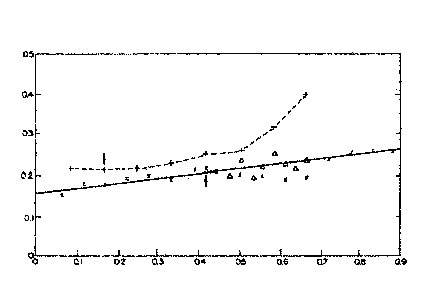
Figure 1: These curves show the effect of waiting time before exposure to microwave energy on final foam density as measured volumetrically in all tests. 20 sec/half power microwave exposure 50 ml material at 75°F. Arrows indicate point where resin gelled at microwave exit.
CROSSLINKED POLYESTER
Highlights of the Technological Achievement: Vesiculated fibrils of crosslinked polyester resin of length 50mm-5 cm diameter 1 mm-2 mm and aspect ratio of 10-50 are prepared by pouring into water with stirring a selected solution in unsaturated monomer of a carboxylated unsaturated polyester resin of acid value 10-90 mg KOH per g, the water containing both a base having a pKb value of 8 maximum and a dispersion stabilizer, and initiating polymerization.
Background: It has been found that one particular group of polyester resins can be used to prepare fibrillar particles of crosslinked resin in which the individual fibril has a vesiculated structure. The term "fibril", means fibrous particles which have diameters of the order of 1 mm to 2 mm and aspect ratios, that is, the ratios of the lengths of the fibrils to their diameters, from 10 to 50. The fibers may be circular or elliptical in cross-section; in the latter case, the "diameter" is considered to be the major axis of the ellipse. The fibril length is typically within the range of 0.5 to 5.0 mm, although by suitable manufacturing techniques this can be extended to about 5 cm.
By a vesiculated structure, is meant that the core of the fibril comprises a plurality of discrete cells or vesicles of liquid or vapor and formed within the body of the fibril. The vesicles vary somewhat in shape, but are usually either spheroidal or elongated, in the manner of a prolate spheroid. An important and distinctive feature is, however, that the internal cells are discrete, isolated entities as distinct from open-ended axial ducts. They are, therefore, not directly accessible to liquids in which the fibrils might be immersed. The vesicle volume of a typical fibril has been shown by mercury porosimetry to be of the order of 45% of the total fibril volume, although fibrils of appreciably lower vesicle volume may be made.
The fibril composition is that of unsaturated polyester resin crosslinked by reaction with ethylenically unsaturated monomer. It is essential that the polyester resin also comprises free carboxyl groups. The preferred concentration of carboxyl groups varies somewhat with the overall composition of the resin, but in general the acid values of useful resins lie within the range of 10 to 90 mg KOH per gram of resin.
It has been discovered that the carboxylated polyester resins which will yield fibrillar particles are distinguished by their performance in the following buoyancy test.
Test - The test is carried out at ambient temperature on a solution of unsaturated polyester resin in ethylenically unsaturated monomer at the ratio of resin to monomer selected to give the desired overall crosslinked resin composition, the ratio being such that the proportion of unsaturated polyester resin in the solution is at least 40% by weight. This solution is hereinafter known as "the test solution."
A droplet of test solution is transferred gently from a probe to the surface of water adjusted to a pH of at least 10 with ammonia. The size of the droplet is not critical, a convenient diameter being 2 to 3 mm. If the test solution is unsatisfactory for fibril manufacture, the droplet will proceed with little if any delay, to drop through the water to the bottom of the container in which it is held. A droplet of test solution suitable for fibril manufacture, will, in direct and obvious contrast, remain suspended on or adjacent to the surface for an appreciable time.
Process Conditions: It has been found preferably to use monomer at least 50% by weight of which is styrene. Poly-functional monomer, for example, ethylene glycol dimethacrylate, is normally used at weight concentrations of less than 10% of the total unsaturated monomer, when a polymer with high crosslinking density is required.
For the satisfactory commercial formation of vesiculated fibrils, the polyester resin must constitute at least 40% by weight of the polyester solution in monomer.
The selected polyester is first dissolved in the ethyleni- cally unsaturated monomer required for the crosslinking reaction.
The polyester solution is then poured into water, in the presence of a base, with continuous mechanical stirring. Discrete fibril precursor particles having the general shape and size of the required fibrils are so-formed.
Example: An unsaturated polyester resin was prepared from the following materials:
| |
Parts |
| Propyleneglycol |
27.11 |
| Fumaric acid |
20.44 |
| Phthalic anhydride |
13.04 |
| Adipic acid |
12.86 |
| Xylene |
3.70 |
The reaction was carried out at 210ºC using the xylene as entraining solvent to strip out water of reaction. The batch was cooled and thinned with 22.81 parts of styrene containing 0.017 parts of hydroquinone, to give a solution of unsaturated polyester resin in ethylenically unsaturated monomer, with the following properties.
| Acid value (calculated on solid resin) |
34.8 mgm KOH/g |
| Gardner-Holdt viscosity |
Y- |
| Nonvolatile content |
68.7% by wt |
A drop of the solution when tested by the buoyancy test described here in above floated at the surface of the water.
A mixture of 6.29 parts of the above solution and 0.11 parts of ethanolamine was poured slowly, with continuous mechanical stirring, into 17.24 parts of water containing 2.26 parts of 0.880 ammonium hydroxide, 2.3 parts of a 7.5% solution by weight in water of poly (vinyl alcohol) and 2.3 parts of a 1.5% solution by weight of hydroxyethylcellulose in water. The grade of poly(vinyl alcohol) used was a partially hydrolyzed (approximately 88%) poly (vinyl acetate) which as a 4% by wt solution in water had a viscosity of 40 cp at 20°C. The hydroxyethylcellulose was a high viscosity grade, which had a viscosity of 1800 cp at 20ºC when tested as a 1% by wt solution in water.
Discrete fibrillar suspended particles of polyester resin solution were formed in the water.
The suspension was diluted with 70 parts of water, and the following materials added slowly and sequentially to it to initiate curing of the particles by a cross-linking reaction between the polyester resin and styrene monomer:
| |
Parts |
| Cumene hydroperoxide |
0.090 |
| Diethylene triamine (as a 10% wt soln. in water) |
0.044 |
| Ferrous sulfate (as a 10% wt soln. in water) |
0.001 |
The cured product was a fluffy suspension of fibrils, the individual fibrils having a diameter of the order of 50 mm and length of 1 to 3 mm. Transverse fractures of the fibrils examined by scanning electron microscope confirmed the presence of a vesiculated structure. The crosslinked nature of the fibrils was shown by their insolubility in warm acetone.
Polyurethanes
FLAME RETARDANCE
Low Molecular Weight Polyurethane Modifier Compounds Yielding Flame Retardance
Highlights of the Technological Achievement: Foamed polymers such as polyurethanes useful, for instance, as cushioning, insulation, and in reaction injection molding to produce thin section moldings can be produced without substantial reduction in physical properties by the incorporation of 2 to 75% by weight based upon the weight of polymer of at least one non-reactive, liquid polyurethane modifier compound processing aid or flame-retardant additive.
Background: Flexible polyurethane foams have been rendered permanently flame-retardant by the incorporation of halogenated glycols such as tribromoneopentyl glycol or dibromoneopentyl glycol in combination with phosphoric anhydride which has been reacted with an alkylene oxide or haloalkylene oxides.
Process Conditions: There are disclosed foamed polymers such as polyurethane compositions comprising the reaction product (a) of at least one polyhydric alcohol with at least one organic polyisocyanate in admixture with (b) a low molecular weight non-reactive modifier compound which is the reaction product of at least one polyisocyanate with at least one monohydric alcohol or at least one mono-functional isocyanate with a stoichiometric amount of at least one monohydric alcohol or at least one monofunctional isocyanate with a stoichiometric amount of at least one polyhydric alcohol or at least one isocyanate-terminated prepolymer with a stoichiometric amount of at least one monohydric alcohol. The disclosed modifier compounds are useful as processing aids or flame-retardant additives for polyurethane foams. Microcellular, rigid, high-resilience, and flexible foam poly-urethanes can be prepared. The foamed polyurethanes can be rendered flame retardant, if desired, by use of a halogenated monohydric or polyhydric alcohol in the preparation of the modifier compound.
The Modifier Compound: The preparation of the low-molecular weight modifier compounds is more particularly described by reference to the following equations:
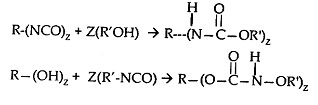
where in R is monovalent or polyvalent and R' is monovalent each individually selected from aliphatic, alicyclic, aromatic, aralkyl, and alkaryl radicals having up to 18 carbon atoms in the aliphatic portion of the radical and 6 to 12 carbon atoms in the aromatic portion of the radical, Z is equal to z and is the number of mols of alcohol or isocyanate compound utilized, and z is an integer corresponding to the valence of R and is equal to the number of functional groups on the alcohol or isocyanate compound. The modifier compounds have a molecular weight of preferably 150 to 900.
Flame-Retardant Polyurethane Foam Prepared by Incorporating a Graft Polymer Having a Particle Size Greater than 0.5 Micron
Highlights of the Technological Acheivement: In the preparation of conventional or high resiliency flexible polyurethane flame retarded foams by reacting organic polyisocyanates with polyols containing vinylic polymers and employing flame-retardant compounds, less flame-retardant compound is required to pass the California Bulletin No. 117 flame test when the particle size of the vinylic polymers is greater than 0.5 micron.
Background: The preparation of flexible polyurethane flame-retardant foam compositions are generally well known as evidenced by the following prior art. U.S. Patent 4,022,718 teaches the preparation of high resilience cold-cured polyurethane foams incorporating 2,3-dibromo-1,4-butanediol as a chain extender and flame-retardant component.
Process Conditions: This process applies to both high-resiliency, flexible polyurethane foam compositions and conventional flexible polyurethane foam compositions which are prepared by the reaction of an organic compound having at least two active hydrogen atoms and having a vinylic polymer content with an organic polyisocyanate in the presence of an effective amount of a flame-retardant compound. The amount of flame-retardant compound required may be significantly decreased if the graft polymer dispersions have a vinyl particle size greater than 0.5 micron.
Among the flame retardant compounds which may be employed are Antiglaze 19, Antiblaze 78, Thermolin 101, FE 55, Firemaster LVT23P, Bromochlor 50, Wiltrof HF, Citex BC26, Fyrol PCF, Fyrol FR2, Fyrol EFF, BCL 462, Phosflex 500, Brominex 161, Firemaster 901, FR 212, FR 1138, Phosgard 2XC20, BC48, Phosgard C-22R, Brominex 214P, Brominex 257, and Fyrol 99.
Low Fire Hazard Rigid Polyurethane Insulation Foam
Highlights of the Technological Achievement: A low fire hazard rigid polyurethane insulation foam having high compressive strength and low friability is disclosed. Rigid foam products of this process have a Class I flame hazard rating according to the ASTM E-84 Steiner tunnel test. The polyurethane foam contains a halogenated base polyol and a modifying amount of a 2,5-bis-hydroxymethyl)furan component.
Background: The fire retardant, or, "low fire hazard" properties of furan ring-containing materials is well known due to the self-extinguishing property of the char which forms when furan-containing materials are burned. The disclosure in U.S. Patent 4,029,61 1 shows a rigid cellular foam having carbodiimide and isocyanurate linkages prepared by catalytically condensing an organic polyisocyanate in the presence of a carbodiimide-promoting catalyst, a trimerization catalyst, and a polyfurfuryl alcohol polymer. The resulting isocyanurate foam has improved flame retardancy. The polyfurfuryl alcohol polymer employed includes the condensation products of furfuryl alcohol with formaldehyde, furfural, urea or mixtures, thereof, produced by reaction in the presence of an acid catalyst providing a pH of preferably 1.5 to 3, at a temperature of 25° to 120°C.
Process Conditions: A preferred hydroxyl composition for use in manufacture of rigid urethane insulating foams, in accordance with this process, comprises 12 to 24% 2,5-bis-(hydroxymethyl) furan component, 76 to 88% halogenated polyol base polyhydroxyl component, as well as 25 to 40 phr (parts per hundred polyol components) blowing agent, 0.8 to 1 .0 catalyst, 1 .5 to 2 surfactant, and 2 to 7 phr of an acid scavenger, such as, for example, an epoxy compound. (Ciba-Geigy CY 179 is eminently satisfactory as an acid scavenger. The latter acid scavenger is 3,4 epoxycyclohexylmethyl-3,4 epoxycyclohexane carboxylate.) This composition is stable for shipment, and can be used as the polyhydroxyl stream in a so-called "one-shot" method, in conjunction with a second, isocyanate stream, to produce rigid insulating foams in accordance with this process.
An example of a commercially available mixture of this type is the material known as FA-REZ B-260 (The Quaker Oats Company). This commercially available product is reported to have a viscosity of about 1 0,000 cp, has a hydroxyl content of 16 to 18%, a water content of 0.5 to 1 .0% furfuryl alcohol content of up to 3% and is reportedly 85% polyhydroxymethyl functionality.
Generally speaking, the polyhydroxymethyl furan mixtures useful in accordance with this process preferably have a viscosity in the range of 6,000 to 12,000 cp at 25°C, contains 35 to 45%, inclusive, monomeric bis-hydroxymethyl furan, less than 3% furfuryl alcohol monomer, less than 1.2% water and have an acid number of less than or equal to 3.5.
Such a hydroxymethyl furan diol mixture which is useful as an ingredient in accordance with this process, can be produced in a number of ways. For example, it can be produced by low acidic (i.e., pH above 4) polymerization of 2,5-bis-(hydroxymethyl) furan, as well as by the hydroxymethylation of furfuryl alcohol with formaldehyde using a weak acid such as, for example, acetic acid, propionic acid, or formic acid, under conditions which provide a pH above 4. Generally speaking, such products can be produced by hydroxymethylation of a furan ring-containing compound selected from the group furan and furfuryl alcohol, wherein the furan compound is contacted with formaldehyde in the presence of a catalytical amount of a weak acid catalyst having a pKa value at 25°C between 3.0 and 5.0 inclusive, under conditions which provide a reaction mixture having a room temperature acidity pH greater than or equal to 4.0, the contacting taking place between 50° and 160°C inclusive.
Intumescent Flexible Polyurethane Foam
Highlights of the Technological Achievement: Flexible, resilient, polyurethane foam having improved flame retardancy and intumescent properties is prepared from a reaction mixture comprising a polyester polyol, an organic polyisocyanate, a blowing agent, a surfactant, a catalyst, a melamine derivative wherein one or more hydrogens have been replaced by a methylol and/or lower alkoxymethyl group, a flame retardant, and hydrated alumina.
Background: Flexible resilient polyurethane foams are made by the reaction of polyols and organic polyisocyanates in the presence of one or more blowing agents, one or more surfactants, and one or more catalysts. The foams find a variety of uses, such as carpet underlay, textile innerlining, mattresses, pillows, furniture padding, cushions, automobile crash pads, and insulation.
Process Conditions: The melamine derivatives used in the practice of this process are those compounds having the formula below.
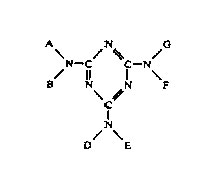
In the preceding formula. A, B, D, E, F and G are hydrogen, hydroxymethyl-(methylol) or ROCH2-, wherein R is an alkyl radical containing from 1 to 4 carbon atoms, such as methyl, ethyl or t-butyl. At least one of A, B, D, E, F and G is hydroxymethyl or ROCH2-. Examples of compounds falling within the scope of the formula are tris-(hydroxymethyl) melamine, tris-(hydroxymethyl)-tris-(methoxymethyl) melamine, hexa-(methoxymethyl) melamine, hexa-(hydroxymethyl)-melamine and tetra-(n-butoxymethyl) melamine.
In general, the amount of compound or compounds of the formula used will be from about 10 to 30 parts by weight per 100 parts by weight of the polyester polyol present in the mixture to be foamed, but greater or lesser amounts can be used without departing from the scope of the process.
The preferred amount is from 15 to 25 parts by weight, per 100 parts by weight of the polyester polyol. The melamine derivatives can be used either as solutions or dispersions in water or other solvents, or as essentially 100% active materials without solvent. It is preferred to use the latter form, in the absence of water, to provide greater latitude in formulating a reaction mixture. When water is used as a blowing agent, it is preferred to add it separately rather than as a solvent for the melamine compound.
A particularly preferred melamine derivative is hexamethy-oxymethylmelamine, which is readily obtainable in a form free, or substantially free, from water. Although it is preferred to use a single melamine derivative, for simplicity in formulating, it may sometimes be desirable to use two or more, and such combinations are included in the scope of the process.
Suitable flame retardants are those conventionally used in the art of making flexible polyurethane foams, and include tri-esters of phosphoric acid, halogenated tri-esters of phosphoric acid, halogenated hydrocarbons, and the like.
The polyester polyol reactants useful in the process include any conventionally used in the preparation of flexible and semiflexible urethane polymer foams. The poly-hydric polyester reactant has a molecular weight of optimally between 500 and 5,000. The hydroxyl number of the compound is correspondingly in the range of 15 to 300. The preferred average hydroxyl functionality for the polyester resins is from 2.2 to 2.8.
Reduced Tendency to Form Embers When Burned
Highlights of the Technological Achievement: Flexible polyurethane foam having reduced tendency to form burning embers when it is ignited and burned is provided by incorporating into the reaction mixture before foaming a ketone or benzalde-hyde.
In a preferred embodiment, flexible polyurethane foam of increased flame retardance is provided by also incorporating a flame retardant into the reaction mixture before foaming.
Background: It is known in the art to add various flame-retardant chemicals to polyurethane foam-forming reaction mixtures, in particular to add halogenated esters of phosphorus. This has resulted in some improvement in the flammability properties, the extent of burning after ignition being reduced and the foams may even be made self-extinguishing to some degree; but while combustion does occur the foam melts and drips flaming embers which may ignite other flammable materials in the vicinity and thus cause the fire to spread. In order to overcome this problem, other additives have been added to polyurethane foam-forming reaction mixtures to render the finished foams intumescent, or to produce a char, once they have been ignitied. Such foams are less prone to the development of flaming, dripping, embers during combustion, and may produce a char which acts as a thermal insulation and thus aids in preventing the spread of a fire.
Process Conditions: This process provides flexible, resilient, polyurethane foam with reduced tendency to form burning, dripping, embers during combustion. These foams are substantially equal in non-drip properties to those provided by the disclosure of U.S. Patent 4,139,501, and are obtained at lower cost. This improvement is provided by adding to a conventional polyurethane foam-forming reaction mixture at least one drip inhibitor such as benzaldehyde or a ketone. The drip inhibitors are generally less costly per weight and generally effective in lesser amounts as compared with the melamine derivatives used in the method of U.S. Patent 4,139,501. The foam-forming reaction mixture also contains a halogenated phosphate ester as a flame retardant.
The products of this process, rather than being stiff and rigid, retain substantially the flexibility, resilience, cell structure, permeability, and hand of conventional flexible polyurethane foams which do not contain the drip inhibitors employed in this process. As a consequence, the foams of this process can be used in most or all of the applications where conventional flexible foams have here to fore been used.
Product: Of the following examples, one of which is a comparative example according to the prior art and others of which are according to this process, those made according to this process are illustrative thereof but not limitative thereof. In these examples, all amounts shown are parts by weight.
The foams were evaluated by a modification of the procedure of UL 94, Standard for Tests for Flammability of Plastic Materials for Parts in Devices and Appliances, published by Underwriters Laboratories Inc. The modification of UL 94 was in the measurement of flame time. The standard test calls for starting to count flame time 60 seconds after ignition, whereas in the procedure used herein flame time was counted from the start of ignition.
The following are the identities of the various ingredients used in the examples. TDI stands for tolylene diisocyanate (also called toluene diisocyanate). The numbers which follow TDI indicate the ratio of the 2,4 and 2,6 isomers (e.g., TDI 65/35 is a 65/35 mixture of tolylene 2,4-diisocyanate and tolylene 2,6-diisocya-nate). A-390 (Witco) is a surfactant comprised of a mixture of modified fatty acid esters plus a silicone; NEM is N-ethylmorpho-line (Jefferson Chemical, Than-cat NEM); DM-16D is n-hexadecyldimethylamine (Lonza, Baircat B-16); NCM is N-cocomorpholine (Lonza, Baircat NCM); C-2 is stannous octoate (M&T Chemicals, Fomrez C-2); Kaydol (Witco) is a white mineral oil; 73D is a dispersion of black pigments in an organic vehicle (black paste); and FR-2 is tris (1,3-dichloropropyl) phosphate (Stauffer Chemical, Fyrol FR-2).
Examples (parts by weight)
| Ingredients |
1 |
2 |
3 |
4 |
| Polyester prepared from diethylene glycol, adipic acid, and trimethylol- ethane; OH No. 56 |
100.0 |
100.0 |
100.0 |
100.0 |
| TDI 65/35 |
50.1 |
50.1 |
50.1 |
50.1 |
| Black dispersion 73D |
7.45 |
7.45 |
7.45 |
7.45 |
| A-390 |
2.0 |
2.0 |
2.0 |
2.0 |
| Water |
3.7 |
3.7 |
3.7 |
3.7 |
| NCM 0.8 |
0.8 |
0.8 |
0.8 |
| Kaydol |
0.2 |
0.2 |
0.2 |
0.2 |
| NEM |
0.5 |
0.5 |
0.5 |
0.5 |
| DM-16D |
0.3 |
0.3 |
0.3 |
0.3 |
| C-2 |
1.5 |
1.5 |
1.5 |
1.5 |
| FR-2 |
20.0 |
20.0 |
20.0 |
20.0 |
| Methyl isobutyl ketone |
- |
5.0 |
- |
- |
| Cyclohexanone |
- |
- |
5.0 |
- |
| Benzaldehyde |
- |
- |
- |
5.0 |
The index of these examples was 111. Example 1 is a comparator, made according to the prior art and not containing a drip inhibitor, and Examples 2 through 4 are according to this process. Each of the examples yielded a flexible, resilient, foam having a uniform cell structure. The flammability characteristics of the foams were evaluated by a modified UL94 procedure, and the results are shown below.
Examples
| |
1 |
2 |
3 |
4 |
| Average time of burn, seconds |
58.7 |
71.9 |
60.5 |
56.3 |
| Average extent of burn, inches |
1.95 |
2.85 |
2.15 |
2.03 |
| Drip formation |
Yes |
No |
No |
No |
| Char formation |
No |
No |
No |
No |
During combustion, dripping embers formed from the product of Example 1 and no char formed. Although the products of Examples 2 through 4 also did not form a char during combustion, they were free from dripping, burning, embers.
Flame Retardant Flexible Polyurethane Foam Containing Finely Divided Inorganic Salt
Highlights of the Technological Achievement: Flexible polyurethane foams which are the product of reaction under foam producing conditions of a combination of: (a) an aromatic polyisocyanate; (b) a polyether polyol with the optional use of a minor amount of a crosslinking polyol; (c) antimony oxide; (d) a polyhalogenated aromatic compound; and (e) a hydrated inorganic salt.
The foams of the process meet very stringent tests for fire retardance required of seat cushioning, mattresses, and like materials in aircraft, institutions such as hospitals, convalescent homes, and the like.
Background: Fire retardant flexible and semi-flexible polyurethane foams which are characterized by good physical properties and good fire resistance are well known in the art; see U.S. Patent 3,909,464 and the art cited therein. The patent discloses the very efficacious combination of antimony oxide, a polyhalogenated aromatic compound and alumina trihydrate. The latter is not actually a hydrate but is rather a true hydroxide.
It has been found that certain hydrated inorganic salts can be employed in place of alumina trihydrate in the fire retardant combination disclosed in U.S. Patent 3,909,464 without there being a problem with excessive or unwanted blowing action in the formation of fire retardant polyurethane foams. In fact, the flexible foams in accordance with this process have physical properties equal, and, in some cases superior, to those of the cited art.
Process Conditions: This process comprises flame retardant flexible polyurethane foams prepared under foam producing conditions from an aromatic polyisocyanate, a polyether polyol having an equivalent weight from 500 to 2,500 and a functionality from 2.0 to 4.0, antimony oxide and a polyhalogenated aromatic compound wherein the improvement comprises employing in the foam forming reaction mixture the following ingredients in parts by weight based on 100 parts of the polyether polyol:
(a) from 4 to 30 parts of antimony oxide;
(b) from 4 to 40 parts of a polyhalogenated aromatic compound; and
(c) from 30 to 60 parts of a finely divided hydrated inorganic salt wherein a mol of the salt compaints at least 5 mols of water of hydration, and employing a ratio of isocyanate equivalents to total the foam-forming reaction mixture within a range of 0.90: 1 .0 to 1.0: 1.0.
Fiber Production
COMPOSITIONS
A variety of polyester compositions have been developed in an attempt to improve fiber properties - particularly dyeability. Litigation has been instituted by Du Pont against Eastman Chemical Products charging infringement of U.S. Patent 3,018,272 issued to Du Pont. That patent involves introduction of sulfonate groups into a polyester polymer chain to give a fiber which can be dyed with basic dyes.
Celanese
A composition developed by R.W. Stockman and D.E. Sargent; U.S. Patent 3,507,835; April 21, 1970; assigned to Celanese Corporation is a composition having improved dyeability properties comprising a synthetic linear thermoplastic polymer and at least 0.5 weight percent of a sulfonated pyrrole.
Du Pont
A process developed by R.E. Taylor; U.S. Patent 3,446,766; May 27, 1969; assigned to E.I, du Pont de Nemours and Co. produces reinforcing yarns and cords of polyester having low free carboxyl group contents which give superior performance in pneumatic tires and other reinforced rubber articles where heat degradation is a problem. A copper salt of an organic carboxylic acid and a molar excess of alkali metal iodide are mixed with fully polymerized, molten polyester to reduce the free carboxyl groups prior to melt spinning. If desired, the metal iodide may be present during formation of the polyester.
Eastman Kodak
A composition developed by H.W. Coover, Jr., and F.B. Jayner; U.S. Patent 3,492,368; January 27, 1970; assigned to Eastman Kodak Company relates to modifications of linear polyesters to provide compositions readily spinnable into fibers and yarns susceptible of permanent dyeing to deep shades by a wide variety of dyes including premetallized and acid wool dyes as well as conventional disperse and other conventional polyester dyes.
Those high molecular weight, fiber-forming polyesters which have met with commercial success in the form of synthetic fibers such as Docron or Kodel polyester fibers are relatively insoluble, hydrophobic materials. Since they are not readily permeable to water, they cannot be dyed satisfactorily by the ordinary inexpensive dyeing procedures. Since they lack reactive groups such as those that abound in cellulosic and protein fibers, they can be dyed satisfactorily only by a limited class of disperse dyes.
Usually it is necessary in obtaining even only light and medium shades to employ high temperature dyeing procedures which are operated at temperatures above 100°C. or with the use of carriers or the use of both high temperature and carriers. Since many dyestuffs decompose at temperatures above 100°C. (e.g., some blues will redden), careful selection of dyestuffs must be made for high-temperature dyeing of polyester yarns and fibers. This markedly limits the utility of these materials in the textile field, because they cannot be dyed readily on conventional commercial dyeing equipment. In addition, carrier dyeing involves considerable extra cost, since (1) the carriers are themselves expensive, (2) the dyed yarns have a pronounced tendency to retain the odor of the carrier and require very thorough washing to eliminate it and (3) residual carrier left in the fiber seriously impairs the light fastness of the dyed material.
The hydrophobic nature of linear polyesters is also manifested in their low moisture regain. The low moisture regain of these materials is conducive to the development of static during processing operations, such as picking, carding, winding and weaving. Although static troubles may be substantially eliminated during processing operations by the application of lubricants and hydrophilic sizing materials, garments produced from linear polyester yarns tend to develop static charges during wear which attract dirt, dust, lint and other materials, thereby causing the fabrics to become rapidly soiled. Furthermore, unmodified polyester fibers, when dyed with the conventional disperse or polyester dyes, have poor resistance to dry cleaning solvents such as trichloroethylene, experience having shown that dry cleaning solvents of this type often extract the dyestuffs from the fibers causing a reduction in shade or producing a mattled effect. As will be more fully set forth hereinÂafter, this process is directed to overcoming these difficulties and to providing polyester fibers and yarns which can be readily and satisfactorily dyed with a wide variety of dyes including premetallized and acid wool dyes as well as conventional disperse dyes.
This process is based on the discovery that certain polymeric blends of fiber-forming linear polyesters with certain condensation copolyamides or homopolyamides can be melt spun or formed into high strength fibers, filaments, and yarns having excellent offinity for premetallized and acid wool dyes as well as disperse dyes and other polyster dyes. The dyed materials obtained thereby exhibit excellent light and gas fastness, and are resistent to the action of prechloroethylene or other dry cleaning solvents.
More specifically the products of this process consist essentially of a chemically heterogenous blend of (A) a linear condensation polyester having an inherent viscosity of at least about 0 .4 and a melting point in the range of about 150° to about 350°C. and (B) a linear condensation polyamide wherein the carbon amide groups are separated by at least two carbon atoms and are an integral part of the main polymer chain, said polyamide having an inherent viscosity of at least about 0.6 and having a melt viscosity substantially equal to or less than that of said polyester.
Societe Rhodiaceta
A process developed by Y. Vaginay; U.S. Patent 3,576,773; April 27, 1971; assigned to Societe Rhodioceto, France involves extruding, in the fused state a branched polyethylene terephthalate containing y/n-2 to z/n-2 equivalent percent of n valent chain units where n is 3 or 4, y is 0.2 and Z is 2 having a specific viscosity of 0.47 to 0 .72 and a melt viscosity of 600 to 2,500 poises and orienting the yarns thus obtained by stretching.
The polymers used to prepare these products are branched polyethylene terephthalates and may be prepared by incorporating a branching agent, having three or four groups capable of forming stable bonds, such as ester, ether or amide bonds with the acid or alcohol groups, into the monomer mixture.
The preferred branching agents are polyols such as trimethylolpropane, trimethylolethone, pentaerythritol or glycerine; polyacids such as trimesic acid, trimellitic acid or anhydride or pyromellitic acid or anhydride; polyphenols such as phloroglucinal or hydroxyhydroquinone; amino acids and acid-alcohols such as hydroxyisophthalic acid or aminophthalic acid; 2,2-bis(4-epoxypropoxyphenyl)propane or diethanolamine.
Depending on the method of polyester synthesis employed 0.2 to 2 mols percent of the trifunctional branching agent relative to the terephthalic acid, or the terephthalic acid derivative, are used. Amounts of the order of 0.1 to 1 mol percent are preferred. The polyesters which may be used in the process are essentially derived from terephthalic acid, ethylene glycol and a branching agent. They can, however, contain a proportion, up to 10 mol percent, of another glycol containing an aliphatic, cycloaliphotic or aromatic group or heteroatoms such as propanediol, butanediol, hexanediol, decanediol, dimethylpropanediol, cyclohexa-nedimethanol, cyclobutanedimethanol, xylylene glycol, polyoxyethylene glycol of molecular weight less than 6,000, polytetrahydrofuran, another diacid such as adipic, sebacic, dodecanedioic, isophthalic or hexahydro or terephthalic acid, or dimeric acids derived from linoleic acid.
These polymers are preferably prepared in the presence of the usual transesterification and polycondensation catalysts and may contain agents to improve their color or heat stability such as phosphorus derivatives e.g., phosphorous acid, phosphoric acid, phenylphosphonic acid or triphenylphosphite. They can also be matted, for example by adding a suspension of titanium oxide.
Fabrics woven from yarns of polyester fibers derived from terephthalic acid and ethylene glycol have exceptional strength, abrasion resistance and pleat retention. However, the development of the use of polyester fibers in loosely woven or knitted fabrics is restricted to a large extent by the problem of pilling. In fact, in these types of fabric the individual fibers tend to migrate towards the surface and to form fibrous globules or pills which cannot become detached because of the high abrasion resistance and tensile strength of the polyester, i.e., the rubbing forces are much lower than the force required to tear the polyester fibers holding the pill.
As it is very difficult to prevent migration of the fibers attempts have been made to lower the mechanical properties of the polyester fibers and to adapt their tensile strength to the rubbing forces applied during normal wear. During wear, the abrasion progressively lowers the tensile strength until it corresponds to the rubbing forces and pill drops off.
It has been attempted to lower the mechanical properties of the polyester fibers using polymers of lower molecular weight. Unfortunately the reduction in the molecular weight of the polyester is accompanied by a reduction in the viscosity in the fused state and in industry it is very difficult to spin a polyester having a viscosity in the fused state at 285°C. of less than 600 poises without the fibers sticking together. It is, therefore, difficult to obtain a fiber suitable for normal commercial use.
Although many attempts have been made to lower the tensile strength by treating the yarn or the woven fabric they have always come up against practically insoluble problems of reproducibility. This provides oriented polyethylene terephthalate fibers which are easily accessible industrially and yarns of fibers which do not give rise to pilling. It provides a polyester-based fiber having an elongation at break of greater than 15% and a tensile strength of 18 to 36 g./tex which comprises a branched polyethylene terephthalate having a specific viscosity of 0.47 to 0.72 and melt viscosity of 600 to 2,500 poises at 285°C. The fibers of this composition may be used in the manufacture of woven or knitted fabrics which substantially do not form pills.
SPINNING
Du Pont
A process developed by D.G. Bennie and T.R. Jones, Jr.; U.S. Patent 3,372,218; March 5, 1968; assigned to E.I, du Pont de Nemours and Company relates to an improvement in melt-spinning artificial fibers. More specifically, it relates to a process for treating the outer face of a spinneret used in the preparation of artificial fibers by the melt-spinning process and to the treated spinneret produced thereby.
The general procedure of forming thermoplastic resins into shapes by forcing them under heat and pressure through an opening generally referred to as an extrusion die is well-known. This procedure of extruding includes a forming of a thermoplastic resin through a specially designed extrusion die known as a spinneret, which is usually referred to as "melt-spinning."
In conventional melt-spinning processes, the spinneret is suspended so as to permit the hot, extruded liquid to fall downward, the extruded streams being cooled and solidified into the filament form during this descent. Among the problems encountered in this process have been flicking, bending, and dripping. Flicking is an obvious and repeated deviation of the liquid stream in the face of the spinneret from the normal path of descent. This phenomenon may occur at regular or irregular intervals. At times, an oscillating effect is observed. When the liquid stream is permanently deflected from its normal path of descent, it is said to have undergone bending. Where the liquid stream has flicked or bent to such an extent that it touches and wets the spinneret face, fouling occurs, the continuous liquid stream is broken, and the liquid thereafter drips from the orifice. This is known as dripping. It has been the practice to wipe the outer face of such spinnerets with a brass rod on a scheduled basis to avoid these problems in so far as is practical.
Several methods have been suggested for treating the face of the spinneret in order to reduce the flicking, bending, or dripping tendencies. This coating was found to be effective for a short period of time but eventually degraded, at the high temperature necessary for melt-spinning of polyesters, and the spinneret then required an even greater amount of wiping until removed from service. An alternate procedure suggested by U.S. Patent 2,403,476, consisting of manufacturing the entire extrusion die from a block of tetrafluoroethylene polymer, has not been found practical for melt-spinning because of inability to machine the small orifices to critical dimensions, particularly non-round orifices.
In accordance with this process, the outer face of a spinneret is coated with a film of an unusually high molecular weight fluorocarbon polymer, by applying a dispersion of the polymer and fusing the coating at considerably higher temperatures, than will be encountered in the melt-spinning process. A film of fluorocarbon polymer having a molecular weight of about 25,000 to 35,000 is particularly effective for overcoming the difficulties mentioned. As a result, the number of spinning discontinuities have been reduced by as much as 70% of the number of discontinuities which result when using one of the techniques of the prior art. In addition, there is no longer any need to wipe spinnerets routinely to remove accumulated deposits. The following is a specific example of the application of the present technique to the improved melt-spinning of polyester fibers.
Example 1: The spinneret is washed with a detergent in water and then dried, inspected, and repaired if necessary. The cleaned spinneret is coated with about 20 mg. of fluorocarbon polymer having a number average molecular weight of about 30,000. The coating is applied by spraying with the described 25% dispersion reduced to half strength with perchloroethylene solvent, using a De Vilbiss spray gun (Model MBC-510-30E) with pressure cup under 25 pounds per square inch (1.8 kg. per sq. cm.) air pressure. The spray adjustments on the gun are set to give a fine mist about the same size as the spinneret diameter. The coated spinneret is placed in a Lindberg Electric Cyclone furnace and held at 370°C. ± 10°C. for 30 minutes. The coated spinneret is then cooled and assembled in conventional melt-spinning assembly. Molten polyethylene terephthalate is then fed to the spinneret assembly at a temperature of 290°C. and under pressure of about 2,500 psi. The extruded melt is spun at a rate of at least 1,500 yards per minute into a 4.35-denier per filament, 600-filament yarn, which provides a'typical 1 ,5-denier per filament yarn after drawing. In a test continuing over a 9-day period using 16 spinning positions, the number of drips per pound of yarn spun is 0 .019.
The following table compares the above run with a run using a lower molecular weight fluorocarbon coating (2), with a run using a silicone coating (3), and with a run using no coating at all on the spinneret (4).
| Example No. |
Coating on Spinneret |
Molecular Weight |
Drips per pound of Spun Yarn |
| 1 |
Fluorocarbon |
30,000 |
0.019 |
| 2 |
Fluorocarbon |
3,700 |
0.176 |
| 3 |
Silicone |
- |
0.038 |
| 4 |
None |
- |
0.18 |
A process developed by N.C. Pierce; U.S. Patent 3,437,725; April 8, 1969; assigned to E.I, du Pont de Nemours and Company relates to the melt-spinning of synthetic polymers and particularly to a method of spinning polymers having higher melt viscosities without polymer degradation and with reduced orientation of the polymer in the spun yarn. It is especially useful in the formation of filaments which have a final denier of less than 50 after they have been drawn.
In melt-spinning of yarns, synthetic linear polymer is melted, forced through the filter of a spinneret pack, then through spinneret capillaries, and quenched to form filaments which are converged into a bundle. The filament bundle is processed to provide the desired physical properties and finally the yarn product is packaged for shipment. Most melt-spinnable polymers are temperature sensitive and degrade rapidly at elevated temperatures. The lower the spinning temperature the higher the pressure required to force the molten polymer through the spinneret. Non-uniform flow, and melt fracture, will occur if the spinning temperature is too low consequently, a compromise is always made between a high spinning temperature for a minimum of polymer degradation.
Another approach to reducing spinning pressures is to increase the size of the capillary. If the same filament size (denier) is desired, this approach requires an increase in the attenuation or stretching of the molten material leaving the spinneret and, ideally, before it solidifies. Unfortunately, this stretching can seldom be done under sufficiently ideal conditions to avoid a spun orientation which adversely affects the final yarn properties. Further, when a heavy filament is spun, the inner portion is not quenched properly due to the insulating properties of the outer portion. This causes non-uniformities across the fiber as well as along the fiber. Frequently, it is necessary to use hot-gas annealing directly below the spinneret to counteract these adverse effects and obtain a more uniform yarn. However, this is costly, both in initial cost and operating costs.
Yet another approach has been tried in which the molten polymer is kept at a relatively low temperature until it is heated as it passes through a heated spinneret which is maintained at a temperature much higher than the melt. In practice, this has not worked because polymer in contact with the top face of the spinneret stagnates, becomes too hot and degrades. The search for better and better yarns goes on continuously. It is now known that improved physical properties, e.g., higher tensile strength, can be provided by polymers with higher melt viscosities, either from high molecular weight polymers or from polymers with stiff chain structures. However, this further complicates the compromise among high spinning temperatures, high spinning pressures, and capillary size. In some instances, it has been impossible to make commercial quality yarns from high viscosity polymers. The process makes possible the spinning of such polymers. For other polymers, it reduces the spun orientation and enables improved properties to be produced in the drawn yarn products.
This process is an improvement over the above processes, and spinneret packs, of the type wherein molten synthetic, linear polymer is supplied to a filterholder at sufficient pressure for the subsequent extrusion into filaments, the polymer is passed through the filter medium in the filter and is extruded through spinning capillaries in a spinneret plate. In accordance with this process, the spinning plate is separated from the filterholder by an insulating medium and spinning capillaries are formed by hollow inserts in the spinnerat plate which provided passages through the insulating medium from the filterholder. The spinning process included (a) passing the molten polymer through the filterholder at initial temperatures within a temperature range below that at which significant polymer degradation will occur, (b) passing the polymer into a plurality of passages each of which leads to a different spinning capillary in the spinneret plate and has an entrance temperature within the initial temperature range, (c) heating the spinneret plate to increase the temperature along the passages from said temperature at the entrance to a temperature at least 60° higher at the spinning capillary, and (d) extruding the polymer from the spinning capillary after a maximum of 4 seconds of travel through the heated passages.
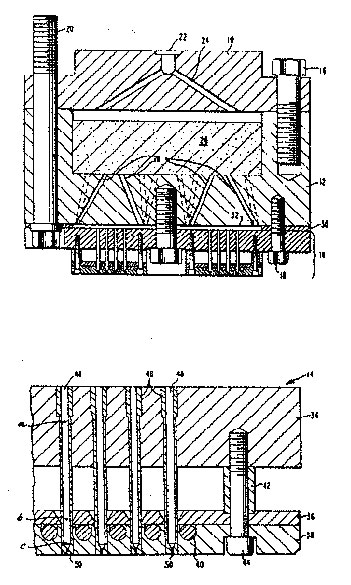
Figure 1 is a sectional view and a detail showing the preferred spinning pack assembly featured in the process. As shown in the figure, lid 10, filterholder 12 and spinneret assembly 14 comprise the main elements of the spinneret pack assembly. Lid 10 is attached to filterholder 12 by a number of bolts 16, while the spinneret assembly 14 is similarly attached to the filterholder by a number of bolts 18. The entire pack assembly is mounted into the spinning machine by bolts 20. Inlet port 22 connects the top face of lid 10 to a plurality of distribution channels 24 which exit on the lower face of lid 10. Sands or screens, or a combination of the two, are placed in cavity 26 in filterholder 12. Distribution channels 28 connect cavity 26 with the downstream face of filterholder 12. Gasket 30, placed between filterholder 12 and spinneret assembly 14, is of sufficient thickness to form distribution space 32.
As shown in the detail in the lower part of the figure, the spinneret assembly 14 includes top plate 34, heating plate 36, and lower plate 38. An electric resistance heating element 40, spirally wound, is embedded in lower plate 38. Spacer 42 provides an air space between top plate 34 and heating plate 36, which acts as a thermal barrier between these two plates. The assembly is bolted together by bolts 44. Hollow inserts 46, one for each filament to be spun, are placed in top plate 34 and extend to the bottom face of lower plate 38. The relatively long tubular passageway 48 of the insert connects the top face of plate 34 to the final capillary 50.
In operation, molten polymer is supplied under pressure by a metering pump (not shown) to inlet port 22 and is distributed equally to the top of cavity 26 by distribution channels 24. After being filtered and sheared by passing through the sand, screens, etc., it is distributed to distribution space 32 by distribution channels 28. Temperature conditions maintained up to this point are such that the temperature of the molten polymer in the distribution space 32 is essentially the same as the temperature of the adjacent metal.
The molten polymer at this temperature is then forced into the top-plate ends of the inserts for spinning through capillaries 50, where the polymer exists in filament shape. Electrical heater 40 supplies heat to maintain the lower plate 38, heating plate 36, and the lower portions of insert 46 at a temperature at least 60°C. higher than the temperature of the supplied molten polymer. Because of the insulation effects of the air space between the plates, very little of this heat reaches top' plate 34, which remains essentially at the same temperature as the molten polymer. Due to the resulting temperature difference, the poor heat transfer characteristics of the polymer, and the extremely short time (a maximum of 4 seconds) that the polymer stays in contact with the high temperature portions of insert 46, a steep temperature gradient is given to the polymer flowing through each insert 46.
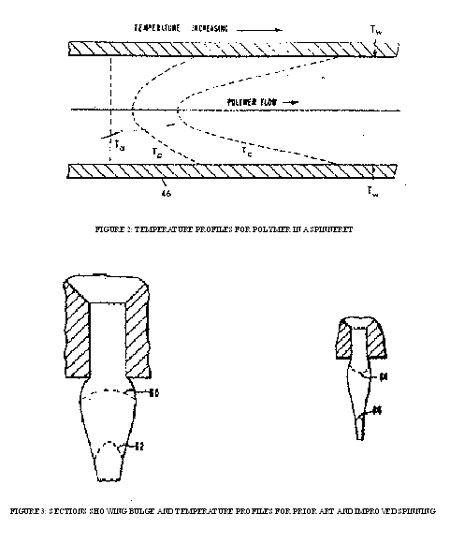
Figure 2. illustrates temperature profiles for polymer flowing through a portion of an insert 46. Initially, the temperature profile across the polymer in insert 46 is essentially constant as shown by Ta (corresponding to point a in the detail of the spinning pack); while at point b, the beginning of a temperature gradient is shown by curve Tb. Just before the polymer enters capillary 50, that is, at point c, there is a steep temperature gradient as illustrated by curve Tc. With a higher temperature on the walls of the entrance hole, the outer layer of polymer has a lower apparent viscosity. Consequently, the pressure required to force the polymer from the entrance hole 48 into capillary 50 and through capillary 50 is considerably reduced even though the temperature of the bulk of the polymer'flowing through capillary 50 has not been increased enough to have an appreciable effect on the polymer.
Figure 3. shows the bulge and temperature profiles for prior art spinning (in the upper view) and for the process (in the lower view). The upper view shows the "carrot" or bulge below a spinneret capillary of sufficient size to spin a high viscosity polymer with a reasonable pressure drop. Line 60 indicates the temperature profile across the polymer just below the capillary. Notice that the temperature of the center portion is slightly higher than that of the outer portion. Line 62 indicates the temperature profile at a short distance from the capillary. Since the exterior of the polymer is being cooled by a quenching medium, the center portion now has a considerably higher temperature than the exterior. It has been found, and is well known in the art, that this temperature profile causes nonuniformities in a filament.
The lower view shows a relatively smaller capillary which can be used with the same polymer when spinning with a heated capillary spinneret according to the process. It illustrates the reduced "carrot" or bulge that results because of less shearing action and less pressure required to force the polymer through the capillary. Furthermore, in order to get the same filament size or denier, less spin stretch is required under these conditions. Line 64 indicates the temperature profile just below the capillary exit. Note that the outer portions of this filament are hotter than the inner portions. A short distance below, line 66 illustrates the profile after some cooling has taken place. Notice that, by this time, the temperature gradient across the filament is very small . This leads to improved product uniformity.
FIGURE 1 : SECTIONAL DIAGRAM OF POLYESTER SPINNING PACK ASSEMBLY
The heated capillary which generates the steep temperature gradient reduces the shear stress at the wall of the capillary without excessive thermal degradation of the polymer. Further, since smaller capillaries can be used without excessive pressure drops, lower spin- stretch ratios can be used to spin low birefringent yarns. In alternate designs, each insert could have a plurality of long entrance holes and capillaries. This is particularly advantageous when the capillaries are arranged in a straight row. Further, it may be helpful to use heat shields or barriers in the air space between the top plate and the lower plate. The following examples are cited to illustrate the operation of the process. The first two examples illustrate the spinning of high molecular weight polymers which cannot be spun with conventional spinnerets. The second two examples illustrate the reduction in spun filament orientation, and improvement in drawn yarn properties; they also demonstrate good spinning without the requirement of hot gas annealing.
FIGURE 2: TEMPERATURE PROFILES FOR POLYMER IN A SPINNERET
Example 1: A series of small batches were polymerized to high molecular weight polymer with the object of preparing fibers with as high a molecular weight as possible (consistent with good processing performance) to achieve superior fiber tensile properties. Inherent viscosities of the various polymer batches for each melt spinning experiment are listed in Table 1. Inherent viscosity ( hinh) is defined as:
where C is the concentration of polymer in the solvent, in g./100 ml. (nominally 0.32 g./100 ml.). The solvent is trifluoroacetic acid/methylene chloride (25/75 vol./vol.). q is the drop time of polymer solution through the capillary viscometer at 25°C. in seconds. q0 is the drop time of solvent through the capillary viscometer at 25°C. in seconds.
Polymer viscosities which could be melt processed by conventional melt spinning techniques had an upper limit of molecular weight of about hinh £ 0.85. Melt fracture occurred at higher molecular weights, preventing continuous spinning. In the examples of Table 1 with conventional spinnerets, a high spinning block temperature of 320° to 325°C. was used, which alleviated melt fracture to some degree but was not sufficient to eliminate the problem. Higher block temperatures degraded the polymer melt. When using the heated capillary spinneret of this method, no melt fracture problems were encountered at a significantly higher molecular weight level. Spun yarn extensibility at high molecular weights and the resulting physical properties were good for the heated capillary spinneret. No direct comparison with conventional yarn could be made because melt fracture prevented yarn processing at these higher molecular weight levels.
Example 2: Poly(bicyclohexyl-4,4'-dimethylene-4,4'-bibenzoate)polymer was produced as disclosed in British Patent 979,401. Its molecular weight was increased by solid phase polymerization. The polymer was heated and spun through a conventional spinneret, and through a heated capillary spinneret of this process. Drawable, smooth yarn is produced with small heated capillaries. With conventional capillaries, large enough to avoid melt fracture, the yarn was essentially undrawable and no low denier-per-filament yarn having good tensile properties could be made. When the test with the heated capillary spinneret was repeated with the capillary temperature reduced to the range of 360° to 375°C., slight melt fracture was observed.
Flex life is defined as the number of flex cycles to failure of the median of single filament samples as tested on a Masland Flex Tester at 70°F. and 62 1/2% RH . Filaments, under a load of approximately 0.33 gpd are flexed over a 0.001 (0.025 mm.) diameter tungsten wire, oscillating through a 180° arc at 150 to 154 cycles/minute.
Example 3: Conventional polyethylene terephthalate polymer melt was spun through a conventional spinneret, and a heated capillary spinneret, to ultimately form a staple product. With the conventional spinneret, the filaments were spun into 360 °C. inert gas in an annealing zone 6 inches long (15 cm.), followed by a delay baffle 16 inches long (41 cm.), a 6 inc (15 cm.) open space, and then to a radial quench unit. The spun yarn was wound up at 1 ,030 ypm (940 meters per min.), at 38 pounds per hour (17 kg. per hour) throughput. Several undrawn yarn bobbins were then combined and drawn on a staple draw machine at 4. OX draw ratio.
With the heated capillary spinneret, a small amount of inert gas (1 to 2 cubic feet per minute) was used to blanket the spinneret face, but no heat was supplied to the gas stream. The yarn was spun through a delay baffle 18 inches long (46 on a 6 inch (15 cm.) open space to the radial quench unit, and wound up at 1 ,030 ypm (940 meters per min.), at 29 pounds per hour (13 kg. per hour) throughput. Two tests were run, with different molecular weight polymers. The products were drawn in identical manner.
Comparison among the test items shows that the yarn from the heated capillary spinneret is much more uniform in diameter and orientation, and could be drawn to a high draw ratio and to superior physical properties without evidence of non-uniformity (e.g. , broken filaments). This was accomplished with the heated capillary spinneret without hot gas annealing.
Using the same heated capillary spinneret as used in Example 3, with the same type of annealing and quenching system for the control, and a small amount of unheated inert gas when operating with a heated capillary spinneret, polyethylene terephthalate industrial yarn was produced.
Three pairs of tests were run, varying the polymer RV and the draw ratio. These data clearly demonstrate that the heated capillary spinneret produces spun yarn with improved properties without hot gas annealing and that a smaller capillary can be used.
A process developed by J.M. Carpenter III and O.K. Smith; U.S. Patent 3,480,706; November 25, 1969; assigned to E.I. du Pont de Nemours and Company provides better filament continuity and a longer spinneret pack life, the improvement including the step of pumping the polymer at a high pressure in excess of 1,000 pounds per square inch gauge through a low pressure drop filter having a pressure drop of 200 to 800 psi to filter the polymer, the filtering being done prior to meter pumping the polymer through the spinneret pack for melt-spinning.
When molten polymer is filtered, agglomerated additives, solid polymer gel and the like are removed and the resultant filtered material is thus rendered more suitable for the production of shaped structures. The production of shaped structures, such as filaments, requires removal of particulate matter that would otherwise clog the exceedingly small spinning orifice. Further, any source of non-homogeneity should be removed so as to reduce accompanying variation in the properties of the filaments. In producing multifilament yarns it is particularly important that the filaments comprising the yarn be as uniform as possible. If, for instance, the yarn contains filaments that vary appreciably in denier or contain segments or markedly different viscosity, subsequent drawing of the yarn will cause objectionable filament breakage.
Figure 4. shows a polyester melt-spinning assembly incorporating such filter devices. Turning now to the drawing, molten polymer in polymer finisher or screw melter 1 is passed to screw pump 3 which forwards the polymer towards booster pump 7 and shear-filter devices 21 and 21' (transfer-line filters) located in transfer-line 5. Booster pump 7, run by motor 9, forwards the polymer through a downstream section of transfer-line 5 to either of shear-filter devices 21 or 21'. When valves 15 and 23 are open and valves 15' and 23' are closed, the polymer will pass through auxiliary line 13 and be filtered in device 21. When it is desired to replace device 21, valves 15 and 23_are closed and valves 15' and 23' are opened to pass the polymer through auxiliary line 13' and device 21'. Flanges 17 and 19, provided for the purpose, are broken, and new device 21 inserted and the flanges resealed. Flanges 17' and 19' are provided for changing device 21' in like manner.
FIGURE 4: SPINNING ASSEMBLY INCLUDING PUMPS, FILTERS AND SPINNERETS
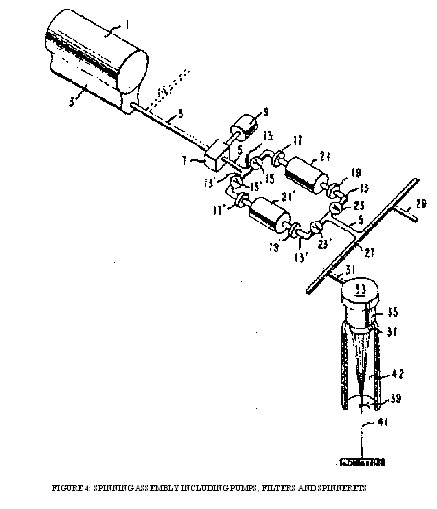
On leaving device 21, the polymer passes to the downstream junction of auxiliary lines 13 and 13' where they unite to continue as transfer-line 5. On passing through transfer-line 5, the polymer travels through header 27 which serves to distribute the polymer to individual conduits such as at 29 and 31 which serve as the supply source of molten polymer for spinning. In spinning, the polymer passes through meter pump 33 in a precise amount and is filtered as it passes through filter 35 and is extruded as small precise streams from a multiplicity of orifices in spinneret 37 and is then quenched in chamber 42 to form the filaments. The filter agent and the spinneret are usually combined and the assembly referred to as a pack. The filaments are converged at guide 39 to form yarn 41. Excess polymer may be diverted as shown by the dotted lines at 4, if desired, to duplicate facilities to those described above.
Figure 5. is a sectional view of the filter used in the process. The assembly comprises an outer shell 53 having inlet 54 and outlet 55. A pipe having plurality of holes 56 which are positioned radially around the circumference and along the length extending into the interior of the pipe. The pipe is mounted concentrically within the interior of the shell. The end of the pipe adjacent the inlet is closed and the other end passes through the shell outlet and forming a fluidtight seal therewith. The pipe is wrapped with layers of various screens 57 to provide the desired filtration. The polymer passes through the screens and into the interior of the pipe and then out of the assembly through the shell outlet.
The following is a detailed example of the conduct of the process in the spinning of polyester fibers. Polyethylene tere-phthalate is polymerized to a relative viscosity of 26 in a polymer finisher, and the molten polymer, which is maintained at a temperature of 285°C., is pumped by means of a screw pump to the inlet of a one-stream gear-type booster pump. The booster pump has a capacity of 16.5 cc per revolution and is capable of delivering 9 to 100 pounds (4 to 68 kg.) of the polymer per hour. The inlet pressure of the booster pump is about 500 psi (35.3 kg./cm.2) gauge and the outlet pressure ranges from about 2,600 to 2,700 psi during test period.
FIGURE 5: SECTIONAL DETAIL OF POLYMER FILTER
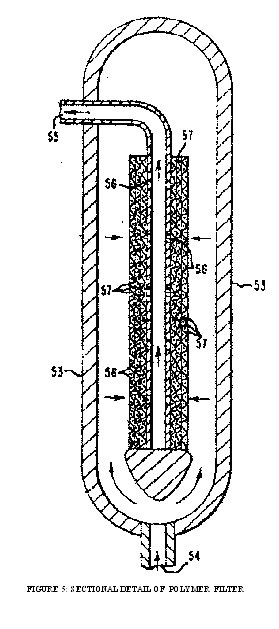
The booster pump forces the molten polymer through a transfer-line filter of the type illustrated. The filter comprises a shell having an inlet and an outlet and a pipe of 3.5 cm. outside diameter for collecting the filtered polymer. The pipe has 8 rows of holes spaced 45° apart aligned along the longitudinal length of the pipe. The longitudinal distance between the holes is about 5 cm., and there are 4 holes per row. The pipe has one end closed and is concentrically mounted within the shell with the closed end being adjacent to the shell inlet and the other end passing through the shell outlet and forming a fluidtight seal therewith. For the filtering medium the pipe is first wrapped with 4 layers of 50 mesh screen (0.023 cm. wires), when 15 layers of 325 mesh screen (0.00355 cm. wires), then 4 layers of 50 mesh screen. The outside diameter of the wrapped pipe is now about 4.5 cm.
The polymer passes into the shell through the inlet and fills the annular space between the shell and the centrally mounted wrapped pipe. The booster pump forces the polymer through the screen wrappings and through the holes of the pipe. The filtered polymer next passes through the pipe and out of the shell through the outlet (via the pipe). The filter area is 580 cm.2 and each hour 11.5 kg. of polymer pass through the filter.
The molten polymer is passed to a header serving eight spinning packs. One of these units contains a pack having a filter consisting of 25 new screens having on effective filtration diameter of 5.08 cm. (19.7 cm.2). The polymer throughput rate through the test pack is 2.88 lb./hr. (1.31 kg.hr.) or 0.145 lb./hr. per cm.2 of effective filter surface. The transfer-line filtration rate per cm. 2 of filtration area is 0.0436 Ib./hr. per cm.2. The ratio of the pack filter rate per unit area to that of the transfer-line filter rate per unit area is 3.33:1. The seven top screens are 20 mesh, the next 16 screens are 200 mesh and the last 3 screens are 325 mesh. The pack also contains a spinneret with 34 orifices having a diameter of 0.038 cm. The system was evaluated for one week. The pressure at the transfer-line filter outlet remained relatively constant at about 2,000 pounds per square inch (141 kg./sq. cm.) gauge. The pressure at the filter inlet increased from 2,430 pounds per square inch (171 .3 kg./sq. cm.) gauge to 2,525 pounds per square inch (178 kg./sq. cm.) gauge over the 7-day test period.
The pressure drop across the pack filter initially was about 875 psi (61.7 kg./cm.2). During the first two days following its installation, the pack pressure increased from 1,830 to 1,890 pounds per square inch (129 to 133.2 kg./sq. cm.) gauge and remained unchanged for the remaining 5 days. When the same test was repeated except that no filter was used in the transfer-line, the pressure increased in a regular fashion from 1,820 to 2,030 pounds per square inch (128.3 to 143.1 kg. per sq. cm.) gauge over the 7-day period. In addition to producing more uniform polymer and filaments of the polymer, the process provides an extended and more uniform pack life.
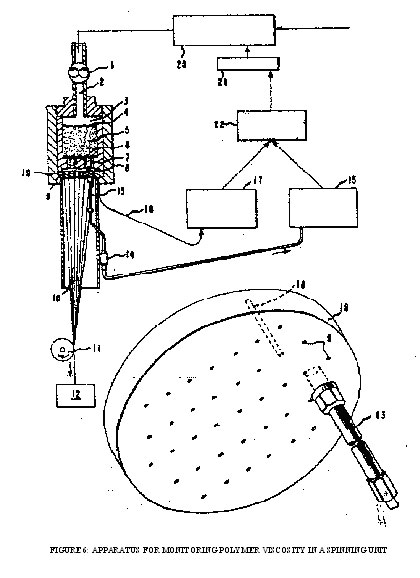
FIGURE 6: APPARATUS FOR MONITORING POLYMER VISCOSITY IN A SPINNING UNIT
A process developed by T.R. Jones, Jr.; U.S. Patent 3,524,221; August 18, 1970; assigned to E.I. du Pont de Nemours and Company involves controlling filament uniformity by measuring the melt-viscosity of the polymer being extruded by measuring the pressure and temperature of the polymer after filtering and just prior to extrusion through the spinneret capillaries, comparing the viscosity with the viscosity desired, and then making the appropriate change in the reactor conditions to change the viscosity toward the desired viscosity.
Figure 6. is a schematic representation of a melt-spinning apparatus fitted with pressure transducer, thermocouple, recording means, and regulator means for correcting the conditions within the polymer finisher for bringing the viscosity back toward midrange together with a detail of a transducer-thermocouple installation in the spinneret plate. As shown in the main view in the figure, molten polymer from a source now shown is supplied to metering gear pump 1 which forces the polymer at a constant rate through conduit 2 into the sand cavity 3 of a melt-spinning head. The polymer traverses the top screen 4, sand filter 5, bottom screen 6 and then moves through channels 7 in the bottom of the sand holder to the space 8 immediately above the spinneret. The polymer is then forced through the spinneret capillaries 9 to form extruded filaments 10 which are quenched, passed over rotating roller 11 and then wound up in package 12.
The pressure of the polymer in space 8 is measured by means of pressure transducer 13, converter into on electrical signal in bulb 14 and indicated and recorded by recorder 15. The temperature of the spinneret is measured by thermocouple 16, with the temperature being indicated and recorded on recorder 17. In normal operation the temperature of the unit is maintained at the desired level by electrical or fluid heating means with the assistance of suitable insulation. The polymer finisher 20 is illustrated as having an associated viscosity regulator 21 which, in the case of condensation polymers, regulates the vacuum with the finisher, thereby effectively controlling polymer viscosity. Integrator and controller 22 determines the actual viscosity as based on the temperature and pressure above the spinneret, compares this with the desired viscosity, and transmits the appropriate signal to regulator 21.
The detail at the base of the figure shows schematically a spinneret plate 19 with a pressure transducer 13 fitted into the face of the spinneret and extending through the spinneret so that the pressure-sensing diaphragm is exposed at the top of the spinneret. Channel 18 is drilled into the body of the spinneret to provide a place for inserting a thermocouple for accurate measurement of the spinneret temperature. The pressure of the molten polymer above the spinneret may be measured with any suitable device of sufficient sensitivity which is stable at the temperature of operation. A suitable device is the Dynisco melt pressure transducer Model PT-422, available from the Dynisco Division of American Brake Shoe. This device uses a bonded strain gauge measuring element for converting pressure indications to electrical signals.
Amplifying and recording devices suitable for use in the process are well known in the art, and a wide choice of suitable instruments is available commercially. A constant rate of flow of polymer through the spinning apparatus may be maintained by any suitable positive displacement pump. Metering gear pumps are preferred.
Once the pressure above the spinneret and temperature of the polymer are known the melt viscosity may be calculated or taken from a previously prepared table based on the constant flow rate and cross-sectional area of the capillaries in the spinneret. The mathematical relationships are amplified by Mc Cabe and Smith in Unit Operations of Chemical Engineering. As before disclosed the process lends itself readily to automatic regulation of the polymer viscosity by controlling the reaction conditions within the polymer finisher.
Integrated Polyester Production Processes
DU PONT
A process developed by J.C. Busot; U.S. Patent 3,487,049; December 30, 1969; assigned to E.I., du Pont de Nemours and Company is a continuous process for producing fiber-forming linear polyester from dimelhyl terephthalate and ethylene glycol. Ester interchange to produce monomer and polymerization of the monomer are carried out continuously in a series of reaction vessels. The addition of a small amount of finely divided terephthalic acid at an intermediate point is shown to increase the degree of polymerization and improve the color under the same conditions, or to increase the rate at which fiber-forming molecular weight is obtained, in comparison with operation of the process without addition of the terephthalic acid.
The manufacturing process starts with the dimethyl ester of terephthalic acid, first carrying out an ester-exchange reaction with glycol at atmospheric pressure to produce the bis-glycol terephthalate, and then heating the bis-glycol terephthalate under reduced pressure to split out excess glycol and form the highly polymeric terephthalate polyester. The reactions are usually carried out continuously in several successive stages in a series of interconnected vessels. The vessels usually include an "exchanger" in which the initial exchange reaction is carried out, a "prepolymerizer" in which low molecular weight polymer is formed at an intermediate temperature and pressure, and a "finisher" where the final stages of polymerization are carried out at high temperature and low pressure. The presence of one or more catalysts is required to provide reaction rates high enough for commercial operation.
An increase in polymerization rate is highly desirable as it allows the use of smaller vessels for a given rate of polymer production, thereby decreasing the amount of capital investment required. Alternatively, an increase in polymerization rate may allow the use of higher pressures in the finisher, and thereby reduce the requirements of the vacuum producing system, or may allow the use of lower temperatures in the finisher with a consequent reduction in side reactions and color formation. Attempts to increase the polymerization rate by raising the catalyst concentration have not been entirely successful. For example, increasing the concentration of antimony oxide, a preferred polymerization catalyst, does not produce a corresponding increase in reaction rate. Furthermore, higher concentration of antimony oxide result in a "graying" of the polymer, presumably from the formulation of metallic antimony in divided form.
This process offers a method of increasing the degree of polymerization in the preparation of polyesters and at the same time providing a polymer having greatly improved color. The process allows higher rates of throughput for equipment of a given size, or for the use of smaller equipment for a given throughput. Other conditions being equal, the process allows for the production of a higher polymer viscosity or for the use of a lower temperature or for the use of a higher pressure in the finisher.
In accordance with this process, it has been found that, in the production of glycol terephthalate polyesters by a poly- esterification reaction carried out at elevated temperatures under reduced pressure, the degree of polymerization obtained is increased by injecting into the low molecular weight prepolymer, undergoing polymerization, a glycol slurry of finely divided terephthalic acid in amounts sufficient to provide from 0.01 to 4 percent by weight of terephthalic acid based upon the weight of polymer produced. Amounts of terephthalic acid smaller than about 0.1 percent by weight do not give an appreciable effect, while amounts greater than about 4 percent limit the degree of polymerization in conventional commercial procedures.
By the term "low molecular weight prepolymer" is meant a polymerizing mixture which has passed the monomeric stage but has not yet reached the desired final molecular weight. Preferably the glycol slurry of terephthalic acid is added to the prepolymer having an intrinsic viscosity in the range 0.1 to 0.5, which corresponds approximately to a 2 to 20 relative viscosity range. Figure 1. is a schematic illustration of the continuous polymerization system used in this process.
FIGURE 1: FLOW DIAGRAM OF INTEGRATED POLYMER SYNTHESIS AND SPINNING PROCESS FOR POLYESTER FIBERS
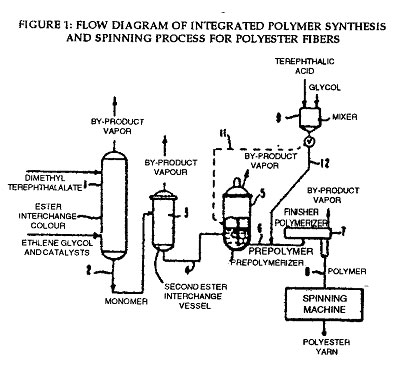
Referring now to the drawing, dimethyl terephthalate, ethylene glycol, and catalyst are supplied to ester interchange column 1 where the exchange reaction is carried out and methanol removed from the top of the column as vapor. Liquid product comprising primarily bis-b-hydroxyethylterephthalate containing a small amount of low polymer and glycol is continuously removed from column 1 through transfer line 2 and injected into the second stage vessel 3 which operates under moderately reduced pressure and elevated temperature to remove excess glycol. Liquid product from vessel 3 is transferred through conduit 4 to prepolymerizer 5 where the pressure is reduced further and the temperature raised sufficiently to give a low molecular weight product having an intrinsic viscosity of about 0.2 to 0.3. This "prepolymer" product is transferred continuously through transfer line 6 to the finish polymerizer vessel 7 where the polymerization is completed and the final product transferred to the spinning machine through conduit 8 for melt-spinning into yarn.
When operated in accordance with this process, terephthalic acid powder and glycol are supplied to mixer 9 for the preparation of a slurry. This slurry is then metered by means of valve 10 either through conduit 11 to prepolymerizer vessel 5 or through conduit 12 to prepolymer transfer line 6, the latter being preferred. It has been found that best results are obtained if the amount of terephthalic acid added to the polymerization system is adjusted to suit the degree of polymerization of the prepolymer at the point of addition. Preferably the amount of added terephthalic acid falls below an upper limit defined by
Z1 = e(1.93 - 0.151RV)
and above a lower limit defined by
Z2 = e - (0.380 + 0.151RV)
where Z is the weight percent, based on polymer, of terephthalic acid added and RV is the relative viscosity of the low molecular weight prepolymer to which the terephthalic acid is added. The concentration of the glycol slurry used in the process may vary over a wide range, but it will be recognized that in a practical commercial process a more concentrated slurry is desirable to prevent overloading the vacuum system with excess glycol. Preferably the slurry is a mixture of terephthalic acid and glycol in a mol ratio within the range 1:1 to 6:1.
This process is found to provide as much as a 50% increase in reaction rate as measured by increased throughput in continuous polymerization systems. The advantages are obtained not only for the production of medium molecular weight polyesters conventionally found in textile grade yarns, i.e., those having an intrinsic viscoisty of about 0.6, but more importantly the advantages are also obtained in the production of very high molecular weight polyesters useful as tire yarns, i.e., those with an intrinsic viscosity of about 0.8 to 0.95. Even when the glycol-terephthalic acid slurry is added in very small amounts which give only a minor increase in degree of polymerization, a marked improvement in polymer color is obtained. The following is a specific example of the conduct of this process.
A continuous polymerization system is used for the production of polyethylene terephthalate of fiber-forming molecular weight. Dimethyl terephthalate and ethylene glycol are supplied continuously to an ester exchanger which is operated in the manner described by Vodonik in U.S . Patent 2,829, 153, issued April 1 , 1958. Sufficient dimethyl terephthalate is supplied to produce approximately 95 lbs. (43.1 kg.) of polymer per hour. Catalysts introduced along with the glycol include 125 parts per million of manganese added as manganous acetate, 50 parts per million of sodium added as sodium acetate, and 510 parts per million of antimony added as antimony oxide. The product of the exchanger is primarily "monomer," i.e., bis-glycol terephthalate with a minor amount of low polymer.
The molten monomer is transferred continuously to a second vessel kept at 240°C. and a pressure of 70 mm. of mercury. A large amount of excess glycol is vaporised and removed in this vessel. The molten material is then transferred to a pre-polymerizing vessel operated at a temperature of about 272°C. and 30 mm. mercury pressure, where low molecular weight polymer is produced. The product of the prepolymerizer, which has a relative viscosity of about 4.5 (a degree of polymerization of about 10), is passed on to a finisher where the temperature is raised to 277°C. and the pressure is reduced to 1.85 mm. of mercury. The final polymerization reaction is carried out in the finisher and the product removed is ready for spinning into yarn.
When the continuous polymerization system described above is operated with a glycol slurry of terephthalic acid (4 to 1 mole ratio), continuously added to the prepolymerizer product at the rate of 2.84 lbs. (1 .29 kg.) per hour (about 1 .20% based on polymer), the final polymer produced has a relative viscosity of 35.2. When no terephthalic acid slurry is added to the prepolymer, the final polymer viscosity is 27 RV. Thus, the addition of terephthalic acid slurry is found to provide a 30% increase in polymer viscosity under the conditions described.
FIBER INDUSTRIES
A process developed by T.C. Higgens; U.S, Patent 3,506,622; April 14, 1970; assigned to Fiber Industries, Inc. is a continuous process for the production of polyethylene terephthalate in which constant equilibrium conditions are maintained in the polymerization stages. Prior to the extrusion stage, excess polymer is removed and treated with ethylene glycol to produce the polymer precursor which is then added to the polymerization reaction at the point of corresponding intrinsic viscosity. The process obviates the necessity for the addition of solid scrap polymer in the final polymerization.
In the commercial production of polymethylene terephthalate polymers, it is highly desirable and feasible to produce large amounts of polymer using a continuous polymerization process. Various methods for the production of polymethylene terephthalate polymers on a continuous basis are known in the art, but these known processes have various disadvantages which can present extensive control problems and provide an undesirable non-uniform quality product. Attempts have been made to maintain a uniform polymer quality in continuous polymerization processes by controlling the level of the molten polymer in the final polymerization reactor by the continuous addition of solid scrap polymer to the monomeric material.
The amount being added being of necessity equal to the increased or decreased demand of the extrusion equipment from the polymer exit. Although it is possible to maintain a satisfactory control of the polymer quality utilizing the addition of solid scrap polymer, obvious disadvantages are present. Of the significant disadvantages of the known continuous polymerization processes, the quality of the scrap polymer to be added must be carefully selected to avoid the addition of undesirable foreign contaminants; the handling problems of the solid scrap polymer are significantly increased and a separate reaction stage is required to convert the solid highly polymeric waste into a liquid low polymer suitable for feeding into the system.
The major disadvantage, however, is in the fact that as the rate of demand for high polymer increases or decreases at the extrusion apparatus; the maintenance of a constant level in the reactor obviously gives rise to variations in residence time which can appreciably affect the degree of polymerization and polymer quality. Further, detection of level changes in the highly viscous polymer in the reactor is difficult instrumentally and the lag or delay between the detection of level changes and adjustment of the quantity of solid scrap being fed upsets the equilibrium conditions in the reactor.
It is the over-all object of this process to provide improvements in the continuous polymerization processes for the production of polymethylene terephthalate polymers wherein variations in supply of polymer to the extruder as required can readily be made while maintaining a uniform quality of polymer throughout the entire reaction process. It is a further object to provide an economic and convenient method for controlling the liquid polymer level throughout the reactor stages to provide a uniform quality polymer with a minimum amount of effort in the continuous production of polymethylene terephthalate polymers.
The objects of this process can be readily obtained in a continuous polymerization process for the production of polymethylene terephthalate polymer which polymerizes bis(hydroxyalkyl) terephthalate by polymerizing at a rate in excess of the maximum requirements and taking off as a side stream the excess polymer produced prior to the extrusion operation in molten state and adding sufficient amounts of the corresponding alkylene glycol containing 2 to 10 carbon atoms used to prepare said bis(hydroxyalkyl) terephthalate to said excess molten polymer to produce the precursor of said polymer. The precursor of the pol-er having an intrinsic viscosity lower than the final polymer can then be returned to the continuous polymerization reaction at the point wherein the intrinsic viscosity of the precursor corresponds essentially to the intrinsic viscosity of the material fed to the high polymer reactor, thus maintaining a constant level of the polymerization mass and at the same time constant throughput and residence time in the reactor. This method provides a unique manner of maintaining invariant conditions within the reactor irrespective of changes in demand for extrusion purposes, eliminates the need for level detection and the considerable upset in equilibrium associated with such methods of control. By virtue of this process modification, therefore the level and throughput of material in the reactor and hence, the residence time are held constant irrespective of extrusion requirements with obvious advantages.
A disadvantage of a continuous polymerization reaction is obvious, however, in that the production of the polymer product must be minutely regulated to the specific needs of the film casting or filament spinning operation. If interruptions occur in the extrusion operation, over production or under production of the polymer can seriously upset the equilibrium of the polymerization reaction. In waiting for the return to equilibrium, valuable periods of time can be lost in the production phase. If reduced extrusion demand suddenly occurs, the reduced throughput in the final polymerizing apparatus, if maintained at constant level of reactions as is taught in the prior art, tends to an increase in residence and reaction time, which can provide a number of undesirable results which include, among others, an appreciably higher molecular weight polymer than desired, producing a non-uniformity of the polymer product.
Additionally, a reduction of the polymer demand can also introduce contaminants in the polymer by increasing the level of the reactant due to relay or lag of the control detecting devices, and the polymer can splash on the upper walls of the reactor and detrimentally degrade. These and other disadvantages can occur. On the other hand, if under production occurs, the polymer to the subsequent metering pumps may be starved and the flow of the liquid polymer to the casting or spinning operation will tend to come in surges instead of in the form of a uniform feed. Such an interruption in the flow of the polymer to the extrusion operation necessitates eventual shutdown of the extrusion operation. In addition, the reduced residence time will cause a lower molecular weight polymer than desired.
A significant feature of this development is to provide a process wherein the deliberate over-production of the polymer which is sent to the extrusion operation can be advantageously utilized. Under these conditions, the excess molten polymethylene terephthalate polymer is removed prior to the extrusion operation. To the excess molten polymer is added sufficient amounts of the corresponding alkylene glycol utilized to prepare the monomeric bis(hydroxyalkyl) terephthalate under reaction conditions which will provide a precursor of the high molecular weight polymethylene terephthalate polymer; such as, monomeric bis(hydroxyalkyl) terephthalate, prepolymer or combinations thereof. The precursor thus produced is returned to the polymeri-zation reaction at the point where the reaction material corresponds essentially to the precursor in composition, in intrinsic viscosity, among other properties.
When the precursor is returned to the polymerization reaction, the supply of the monomeric bis(hydroxyalkyl) terephthalate is diminished or cut off in amounts to correspond to the amount of precursor added. Under these conditions, the level and flow rate and residence time of polymerizing material in the reactor is maintained substantially constant providing at all times a polymer of the desired uniform composition to the extrusion operation and irrespective of the change in demand. One feature which is indeed surprising relates to the absence of detrimental degradation, e.g. the production of contaminants, of the high molecular weight polymer in its reaction with the added alkylene glycol to produce the precursor which in turn can be further polymerized to obtain the desired high molecular weight polymer product. Figure 2 is a flow diagram showing the essential features of this process.
For the production of bis(2-hydroxyethyl) terephthalate from terephthalic acid and ethylene glycol, a direct esterification reactor 10 containing a terephthalic acid inlet line 11 and an ethylene glycol inlet line 12 is maintained at temperatures in the range from about 200° to about 275°C. The amounts of ethylene glycol and terephthalic acid added to the reactor can range from about 1.0/1.0 to about 3.0/1.0 molar ratios of glycol to terephthalic acid, respectively. The esterification reaction conditions are continually maintained, to produce the initial monomeric bis(2- hydroxyethyl) terephthalate products; such as, half esters, esters, dimers and the like. During the esterification reaction, the volatiles which form, such as water, are removed through exit line 13. Monomeric bis(2-hydroxyethyl) terephthalate product produced in reactor 10 is then passed through line 14 into the prepolymerizer 15 wherein the initial polymerization reaction occurs at temperatures in the range from about 250°C. to about 285°C. and at pressures in the range from about 5 to about 60 millimeters mercury.
During the prepolymerization reaction, the ethylene glycol removed can be recycled through lines 16 and 25, back to the source of the original ethylene glycol for further use without an additional purification step in the esterification. The prepolymer formed having an intrinsic viscosity in the range from about 0.1 to about 0.4 is then passed through line 17 into the final polymerizer 18 which is maintained under similar conditions as the prepolymer reaction but generally lower pressures in the range from about 0.5 to about 10 millimeters of mercury. As the prepolymer is passed through the final polymerizer, a more complete polymerization occurs, i.e. the intrinsic viscosity of the product increases from about 0.4 to about 1.1, or higher, progressively as it passes through the final polymerizer. The product of the final polymerizer is withdrawn when the desired intrinsic viscosity of the polymer is obtained or conditions can be adjusted to obtain the desired product out of the exit line 21 which can be passed on to an extrusion operation, such as spinning into fiber or filament production.
Assuming a sudden reduced demand of polymer rate to the extrusion apparatus, the excess amount of polymer produced can be withdrawn from line 2l through line 22. Line 22 is heated not only to maintain the excess polymer in a molten state but also to provide sufficient heat for the reaction of the added ethylene glycol to form the precursor described hereinafter. It is not desirable to maintain the excess polymer in its molten state over extended periods of time since contamination degradation can occur.
The excess molten polymer in this process can be treated in several ways for further use. The ethylene glycol which is produced in the final polymerizer can be added via lines 19 and 20 to the excess polymer in heated line 22 to produce the polymer precursor, i.e. prepolymer, monomer, or mixtures thereof. If sufficient ethylene glycol is recovered from the final polymerizer to produce the presursor equivalent in composition to the prepolymer in vessel 15 or line 17, the precursor can then be returned via line 22 through line 24 to vessel 15 and returned to the final polymerizer through line 17 for further polymerization. If the amount of ethylene glycol from the final polymerizer in line 20 is not sufficient, additional ethylene glycol can be supplied to line 20 or line 22.
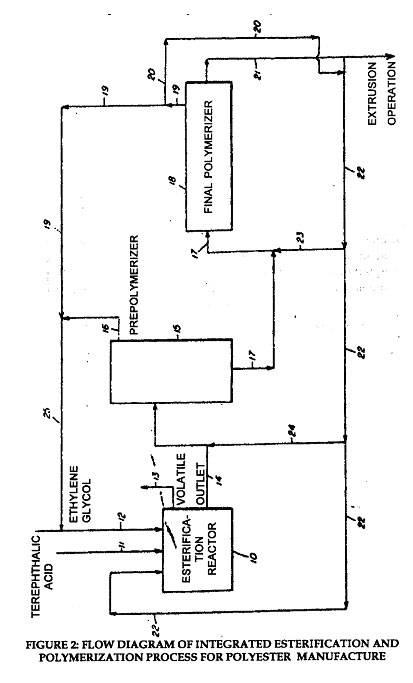
A method of determining composition of the precursor in line 22 can be made by a measurement of intrinsic or melt viscosity of the precursor. It should be noted at this point, however, that if the precursor is added back to the final polymerizer, the amounts of prepolymer or monomeric material prepared prior to the final polymer should be diminished by adjusting the flow of materials fed via lines 11 and 22 and also 12, if desired, in amounts corresponding stoichiometrically to the amounts of the precursor added to the final polymerizer. Under these conditions, the desired process parameters in the final polymerizer will be maintained thereby assuring a uniform polymer product and avoiding production of undesirable contaminants. Additionally, if the precursor produced in line 22 is equivalent in composition to the monomeric material produced in the esterification reaction and supplied through line 14, the precursor produced in line 22 can be added through line 24 to the prepolymerizer, if desired, with the appropriate change of reactants via lines 11 and 22 mentioned above.
The highly perferred procedure is to transfer the excess molten polymer in line 22 to the esterification reactor 10 and ad-justing the supply of terephthalic acid and glycol. Under these conditions, control of the operating process parameters, particularly flow rate, level and residence time of the reactants in all units of the process are maintained invariant. The amounts of ethylene glycol added to the high molecular polymer to produce the precursor can be made, if desired, equivalent on a molecular basis to the amount of ethylene glycol needed for reaction with the terephthalic acid fed via line 11 to produce the monomeric bis(2-hydroxyethyl) terephthalate product. Under these conditions, the ethylene naturally required and fed to item 10 via line 12 could be discontinued.
Alternatively, no glycol at all need be added to the molten polymer in line 22 but could be transferred back to the esterification reactor via lines 19 and 25. The excess high molecular weight polymer passing via line 22 would not be reacted with glycol in route but would then be transformed to the essentially monomeric bis(2-hydroxyethyl) terephthalate in the esterification reactor where it is reacted in situ with the glycol via line 12, and the product would be used for further it polymerization, as described herein. As can be readily determined, there are many additional variations for the return of the polymer back to the polymerization process which are inherently incorporated in the description of the accompanying drawing.
Although the description of the drawing specifically includes the use of terephthalic acid as one of the starting materials, dialkyl terephthalates such as dimethyl terephthalate can be utilized in the preparation of monomeric bis(hydroxyalkyl) terephthalate products. In the preparation of the polymethylene terephthalate polymers by this process, minor amounts of known modifying materials, if desired, may be added, i.e. various glycols, various dicarboxylic acids, dye modifiers, catalysts, inhibitors, and the like. These modifiers may be added as one of the initial reactants during the esterification and polymerization process or the modifying material may also be polymerized separately and melt blended with the polymethylene terephthalate material desired.
In addition, and without detrimental effects to process, various additives, such as delustrants and the like, can be added to the polymer product to obtain desired effects in the polymer product as well as catalysts to increase the speed of reaction in the method of this process.
GOODYEAR TIRE AND RUBBER
A process developed by B.W. Pengilly; U.S. Patent 3,427,287; February 11, 1969; assigned to Goodyear Tire and Rubber Company involves the production of linear polyesters by reacting a free dicarboxylic acid with a glycol in a solvent which is a low molecular weight linear polyester to produce a low molecular weight linear polyester product having an average degree of polymerization of from 3 to 20. This low molecular weight polymer can then be polymerized by condensation reaction. The process can be advantageously operated as a continuous process by continuously adding free dicarboxylic acid and glycol to molten low molecular weight linear polyester resin, reacting them and continuously withdrawing an amount of low molecular weight polyester resin equal to the acid and glycol added. High molecular weight linear, polyesters can be produced continuously by continuously adding free dicarboxylic acid and glycol to molten low molecular weight linear polyester resin and reacting them while continuously withdrawing low molecular weight resin and introducing the resin withdrawn into a polymerization apparatus and continuously polymerizing the resin to high molecular weight resin and withdrawing high molecular weight linear polyester resin from the polymerization apparatus. Figure 3 is a flow diagram of this process.
In operating continuously, to start the process, heated reaction vessel 1 is filled about one-third full with low molecular weight polyethylene terephthalate. Then this low molecular weight polymer is heated to a temperature of from 250° to 260°C. and slowly agitated by means of stirrer 2 attached to shaft 3 which is turned by motor 4. A storage vessel 5 is used to store a supply of terephthalic acid. Terephthalic acid is conducted from storage vessel 5 by means of a screw conveyor 6 through conduit 7 into the heated reaction vessel 1 . A storage vessel 8 is used to hold ethylene glycol. Ethylene glycol is conducted from the storage vessel by means of control valve 9 through conduit 10 into heated reaction vessel 1. The terephthalic acid and ethylene glycol are run in continuously and simultaneously in approximately stoichiometric proportions. Water released in the esterification reaction is vaporized. Water vapor together with ethylene glycol vapors pass into reflux condenser 11 where the ethylene glycol is condensed and returned to the reaction vessel 1.
Water vapor is drawn off through line 12 at the top of condenser 11 and discarded. Low molecular weight polyester is formed and is withdrawn from heated reaction vessel 1 at about the same rate that the reactants are introduced into heated reaction vessel 1 by means of control valve 13 through conduit 14 and conducted to polymerization vessel 15 in which the low moleular weight polyester resin is passed in at one end, polymerized while passing through the reactor and the high molecular weight resin formed is removed from the other end. Polymerization catalyst, stored in storage vessel 16, is continuously conducted through control valve 17 through conduit 18 into polymerization vessel 15. Water vapor and glycol released in the polymerization reaction are withdrawn from the polymerization vessel through condenser 19 and passed to glycol recovery unit 20 where the glycol is recovered.
FIGURE 3: FLOW DIAGRAM OF ALTERNATIVE INTEGRATED ESTERIFICATION AND POLYMERIZATION PROCESS FOR POLYESTER MANUFACTURE
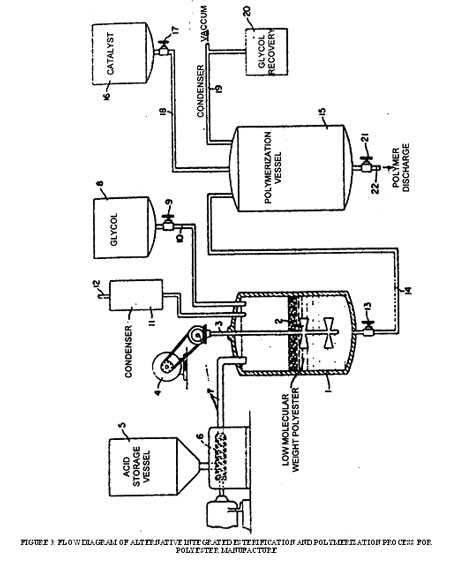
The polymerization vessel is operated under reduced pressure by a vacuum system, not shown, which is connected to condenser 19. High molecular weight polyester resin is removed from the polymerization reaction vessel through valve 21 and conduit 22 and sent to another station not shown where it may be stored or processed into fiber, film, or other products.
This process has many advantages, both technical and economical. The free acids are less expensive than dialkyl esters of acids so considerable savings can be made by using the free acid where the prior art processes use the dialkyl esters. There is no byproduct of lower alkyl alcohol and since the excess of glycol used is kept at a minimum, recovery and losses of glycol are considerably reduced. Reaction rates are rapid and complete reaction from raw material to high polymer may be carried out in as little as three hours. Futhermore, polyesters formed by this method have intrinsic viscosities which are somewhat higher than those normally obtained by the ester interchange route.
In addition to these advantages, the polyester product may contain much less catalyst residue than polyester resin formed by the ester interchange process. No catalyst is needed in the initial esterification reaction although catalysts such as zinc acetate, manganous acetate, and alkali metal alcoholates may be employed if desired. The only catalyst actually necessary is a polymerization or condensation catalyst which may suitably be a material such as antimony trioxide, zinc borate, litharge, lead acetate, magnesium oxide, or other condensation catalyst. In addition, stabilizers such as phosphates and phosphites can be added during the process without any adverse effect on the polymerization reaction which is contrary to the effect of such materials when they are used in the ester interchange method.
MONSANTO
A process developed by E.J. Dobo; U.S. Patent 3,402,023; September 17, 1968; assigned to Monsanto Company involves an apparatus for the continuous processing of linear polycondensates wherein a single column houses the entire system, which column may be operated at a single temperature condition throughout. The various functions incident to a typical polycondensation process are carried out within the present column while employing substantially no moving parts and is so designed as to be generally applicable to linear condensation polymerization systems in general insofar as they involve the typical steps of (1) true monomer formation, (2) evaporation of volatile by-products, and (3) finishing or final polymerization.
Such a column, when disposed in operative position, would extend vertically and consist of three major zones, viz. reactor, evaporator, and finisher zones, in descending order. Such process and apparatus is susceptible to heretofore economically unattainable miniaturization, thereby rendering it feasible for laboratory-scale operations, while at the same time capable ,of affording substantial savings in capital investment in large-scale commercial operations. The finisher section of this polyconden-sation apparatus is characterized by a series of superposed stages which are level controlled by a simple device which automatically functions to regulate the level in each stage in a manner whereby each stage level is independently maintained below a predetermined maximum level. This is accomplished by varying the inter stage gas discharge area to thereby vary the interstage pressure drop, resulting in a liquid flow variation inverse to the drop, resulting in a liquid flow variation inverse to the variation in interstage gas discharge area. Figure 4 shows the assembled apparatus as well as specific constructional details.
The polycondensation system 10 is seen to be in the form of a vertically extending, horizontally partitioned column 11 defining a reaction zone, an evaporation zone and a finisher, or final polymerization zone, indicated by brackets labeled A,B, and C respectively.
The reactor zone or chamber A is essentially a vertical column of a number of discrete stages 12. The output of one reaction stage becomes the input of the successive stage therebelow. Simple standpipe overflow controls 14 operates to individually control the level and therefore the volumes of the reactants in the various stages, while at the same time, providing a convenient vapor outlet. The reactor stages 12 may be defined by simple buckets 16 suspended centrally by a common suspension rod or cable 18, the standpipes 14 preferably being formed integrally with the bottom of such buckets and communicating therethrough to discharge into the next lower bucket.
Alternatively, such reactor stages may as well be defined by partition plates positioned substantially equidistant along the length of the reactor zone, which plates may be wedgingly engaged with the internal wall of the column 11 . The column may be constructed of any suitable non-reacting material, such as stainless steel or glass, the only criterion being that it have sufficient structural integrity to withstand anticipated reactor pressures, depending upon reactant properties and the polymer to be obtained. For example, the reactor zone may be operated above 75 psig in a polyester system.
The design of the reactor for use in a polyester system is based on simple kinetics. By increasing the operating temperature of the reactor section, it is found that one can enormously increase the reaction rate by increasing the specific reaction rate according to the general Arrhenius equation. By allowing the pressure of the by-products, (methanol in the case of an ester interchange between dimethyl terephthalate and glycol) to build up over atmospheric pressure, the operating temperature of the reactor can be raised to,a suitable level. In addition to increasing the kinetic rate in the ester interchange process, the relative volatility of the reactants with respect to the by-products may be considerably decreased, thereby retarding loss of chemical and minimizing the adverse effects of possible sublimation.
Another advantage of operating the reactor section at substantial superatmospheric pressures lies in the fact that higher enthalpy may be imparted to the reacting medium due to the higher temperatures that may be utilized by virtue of such high pressures which increased enthalpy is of advantage in subsequent operations, viz. evaporation. As will be further related too, this increased enthalpy makes it possible to flash vaporize most of the excess chemical (ethylene glycol in the case of the polycon- densation of polyethylene terephthalate; excess water in the cases of nylon) on passing from the reactor zone A to the evaporator zone B.
When the reactor is operated under pressure, a pressure gradient is generated between the reactor A and the evaporator B, the latter preferably being operated at atmospheric pressure or below. Consequently, it is possible to subject the fluid departing the reactor to a throttling process. This is partially accomplished by a simple, internally-contained reactor zone level control 28, in the form of a spring-biased, float-actuated valve, shown in greater detail in the view at the upper right of the figure, which valve operates to control the level at the bottom of the reactor section A to thereby constitute an inter-zone pressure seal between the reactor and evaporator zones and prevent pressure blow-out from the reactor into the evaporator. The reaction mixture, which, in the case of polyethylene terephthalate, is in the form of bis(2 hydroxyethyl) terephthalate monomer and excess ethylene glycol departs the reactor zone A to pass to a spray nozzle 20, where the remaining portion of the throttling process takes place to vaporize most of the remaining ethylene glycol.
FIGURE 4. SINGLE TOWER REACTOR DESIGN SUITABLE FOR INTEGRATED ESTERFICATION AND POLYMERIZATION IN POLYESTER MANUFACTURE
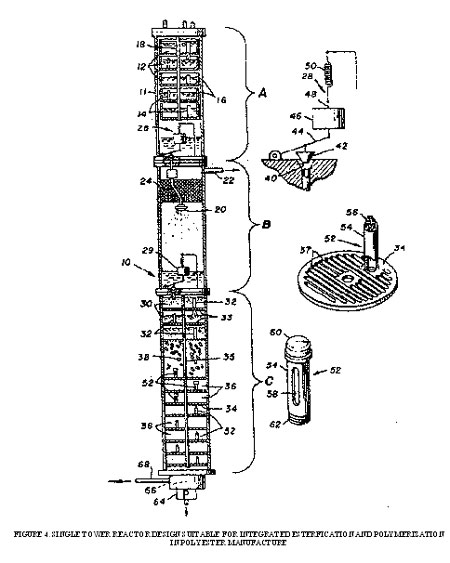
The throttling process involves a condition of constant enthalpy wherein the sensible heat of the liquid fed from the reactor zone is converted into the latent heat necessary for flash evaporation of the excess chemical, which excess is led off through vent 22, after having circulated through demisting material 24, which functions to entrain non-vaporized chemical that might otherwise be carried off with the relatively volatile by-products. It will be appreciated that the evaporation ratio due to throttling increases with increasing feed temperature and with decreasing pressures in the evaporator section. In the preferred operating range, for the processing of polyethylene terephthalate wherein the entire system is maintained between 250° and 275°C. and the reactor section is maintained at approximately 75 psig, the high enthalpy of the reactor discharge is sufficient to flash vaporize most of the excess chemical in the immediate vicinity of the spray nozzle 20.
The discharge from the spray nozzle 20 in the evaporator zone B is preferably of hollow-cone-shaped configuration, whereÂby the reaction mixture is caused to impinge against the heated interior surface of the evaporator, resulting in what is essentially a falling-film evaporation process, which process nicely complements the evaporation due to throttling. The higher heat transfer coefficient obtained in a falling-film evaporator is largely due to the fact that much greater linear velocities are obtained for a given weight of fluid than would overwise obtain if the tube were substantially full. This falling-film evaporation process supplies the additional heat which may be found necessary to insure complete evaporation of the remaining excess chemical if any although it is quite possible that either the falling-film or throttling action may be found self-sufficient under certain process conditions.
The evaporator zone B can be operated under a vacuum as well as under substantially atmospheric conditions, keeping in mind that the degree of polymerization of the prepolymer produced in the evaporator is dependent on the pressure maintained in such zone. The prepolymer product, in the form of bis-2-hydroxy-ethyl terephthalate or low molecular weight polyethylene terephthalate, or mixtures of the two, depending on process conditions, leaves the evaporator zone by way of a simple evaporator zone level control 29 substantially identical to reactor zone level control 28. This control 29 functions as a pressure seal on the prepolymer fluid passing from the relatively higher pressure of the evaporator zone B to that of the high vacuum maintained within the finisher zone C.
Referring now to the details of the structural features and operation of the finisher zone or final polymerizer C, it will be appreciated that, in the normal condensation polymerization process, a small molecule is eliminated as a reaction byÂproduct. This reaction by-product must be removed rapidly and continuously to allow the final polymerization reaction to proceed efficiently. This necessity for efficient reaction by-product removal is complicated by the fact that, as polymerization of the reacting fluid progresses, there is an accompanying increase in its viscosity, with the result that, in the earlier stages of the finisher or final polymerizer, the controlling rate regarding by-products evolution and removal is one of simple kinetics; whereas, as the reacting fluid advances toward the outlet end of the finisher, the controlling mechanism shifts to what may be characterized as that of diffusion. The problem presented by this shifting of the controlling mechanism due to increasing viscosity is compounded by the further problem encountered in regulating the fluid levels of the various stages of a vertical flow type finisher, such level control being essential to provide the desired hold up of the reacting medium at the various levels within the finisher. The present finisher is designed to answer both of these problems in a most simplified and effective manner, as well as offering a convenient exhaust route for glycol vapors.
It is seen that the finisher zone C, in its upper stages, bears strong resemblance to the reactor stages 12 of the reactor zone A. Because of the lower viscosity at the earlier stages of the finisher and because of the high rate of gaseous evolution of reaction by-product, which evolution serves to agitate the contents of the upper finisher stages, the mode of such earlier stages is that of stirred-tank stages 30. These upper-finisher, stirred tank stages 30 are each provided with simple standpipes 32 for stage level control purposes, these standpipes being of much the same construction as that of the reactor stage standpipes 14 employed in the reactor zone A. The upper finisher stages 30 may be defined by any suitable means such as buckets or plates, such as the imperforate partition discs 33, which discs may be wedgingly engaged against the wall of the finisher zone, or suspended at fixed points along a suspension rod 35.
In the intermediate and lower stages of the finisher zone C, the mechanism limiting the rate of by-product vaporization begins to shift from that of simple kinetics to include that of diffusion due to the increasing viscosity of the reacting medium. It therefore becomes necessary to modify the structural characteristics of these intermediate and lower finisher stages in order to generate the fluid surface and renewal necessary for efficient diffusional operations. Accordingly, these stages are defined by perforated partition discs 34 or buckets having perforated bottoms to thereby define what may be termed "plug-flow" type reactor stages 36.
Where the partitioning means takes the form of a disc, such disc is preferably in the form of a turbogrid plate 34, best seen in the detail at the middle of the right hand side of the figure. Such a plate or disc has formed therein substantially equispaced, chordwise extending slots 37, which slots are operative to generate the necessary surface renewal within the relatively more viscous fluid medium passing therethrough. In certain polycondensation processes, wherein relatively lower or intermediate ranges of viscosity are encountered, it may be found desirable to provide an additional surface renewal agency in the intermediate finisher stages in order to promote more efficient by-product evolution. Such a surface renewal agent may take the form of conventional packing 38, such as Raschig rings or Berl Saddles, which 'packing may occupy substantially the entire volume of the intermediate stages. The number of intermediate stages which may beneficially employ the additional surface renewal agency in the form of packing is best arrived at by trial and error and is indicated when the viscosity of the reacting medium has reached such a level that an open-coated flow over the individual elements does not obtain. From this point downward the remaining finisher stages would more efficiently employ only the slotted plates 34 or sieve trays as a surface renewal agency, which plates, by virtue of the viscosity there obtaining, will serve to hold the reacting volume at each of the lower stages the desired period of time and which plates or trays will of themselves, create the surface renewal necessary for diffusional operations, which are now paramount, to take place. It, of course, follows from the present discussion that, in polycondensation systems where the final level of viscosity is relatively low, it may well be found desirable to provide even the final stages with the packing otherwise employed only in the intermediate stages.
Attention is now directed to the detail at the upper right of the figure, wherein there is illustrated the details of one possible embodiment of the zone level control mechanisms 28, 29 employed in the lower ends of the reactor and the evaporator zones A, B. respectively. Reference will be had to only one possible construction of such a zone level control mechanism in that the controls for both the reactor and the evaporator zones may be of substantially identical construction. Such a control will preferably be in the form of a spring-biased, float-actuated proportional valve having a tapered valve seat 40 shaped to receive a similarly tapered valve number 42.
By token of such a configuration, the discharge area through the valve varies according to the displacement of the valve member. The valve member 42 freely depends from pivoted lever 44, the free end of which is hingeably connected to float 46 by means of the resiliently suspended float rod 48. The resilient suspension may be in the form of a tension spring 50 depending from a fixed mounting. The spring suspension serves to render the float 46 and its associated linkage substantially weightless, whereby any variation in the level of fluid in which the float is suspended will be precisely translated into a vertical displacement of the float. For example, a rise in the position of float 46 will be translated into a rise of the valve number 42 relative to the valve seat 40 to thereby define an increasingly large area of discharge through the valve.
Turning now to another significant feature of the apparatus, there are shown in the two lower detail views, two possible embodiments of a simplified finisher stage level control 52 designed to be employed in the intermediate and lower finisher stages, insofar as such stages employ perforated discs or plates 34, or sieve trays, wherein the interstage fluid flow takes place through such discs, as opposed to the upper stages, wherein the interstage flow takes place by way of upper finisher stage standpipes 32, similar to the reactor stage standpipes 14. The features of the finisher stage level controls constitute one possible construction which may be employed in the practice of the method of individually controlling the liquid level within various stages of a vertical-flow finisher, which method is based on the concept of controlling the rate of interstage liquid flow by generating variations in interstage pressure drop, which variations are accomplished by varying the area of interstage gas discharge to thereby maintain the liquid level within a given finisher stage below a desired maximum.
One way of accomplishing this variation of interstage gas discharge area is to provide an interstage gas passage independent of the interstage liquid passage, which passage is so designed that its cross-sectional area is subject to a controlled variation induced by variations in the liquid level of given stage, which variations in area induce variations in the interÂstage fluid pressure drop.
For example in one embodiment illustrated such a finisher stage level control 52 may be in the form of a cylindrical body portion 54 the lower end of which sealingly engages disc 34 and communicates there through to register with the next succeeding finisher stage below. The upper end of body portion 54 is enclosed by a tapered cap portion 56 which is preferably constructed of a substantially fluid-impermeable-gas-permeable material such as small-mesh, stainless steel screening. The dimensions and configuration of the cap portion 56 may vary according to system capacity and the conditions to be obtained, the only essential criterion being that such cap portion vary in cross-sectional area in a direction parallel to the axial dimension of the body portion 54, viz., the cap portion cross-sectional area will vary in a vertical direction.
In operation, liquid flow from an upper stage will collect upon and flow through the slotted disc 34. Should the liquid flow through the slots become less than the rate of liquid discharge into a given stage, the liquid level will rise along the body portion 54 to a point of maximum desired height, whereat it will begin to engage the tapered, gas-pervious cap portion 56. Up until this point, the pressure drop between the stage being considered and that stage next below has remained constant due to the fact that the area of gas discharge through the body portion has remained constant. As the liquid level rises along the cap portion 56, a progressively smaller area of gas discharge therethrough is defined, with the result that there is an incremental increase in pressure within the stage under consideration. It will be appreciated that, if upon such an increase in pressure within one stage relative to another, the fluid flow therebetween by way of the perforated disc 34 will increase.
If such increase in flow is insufficient to arrest or reverse the rise in liquid level, a still smaller gas discharge area will be defined to thereby generate still larger pressure differentials and concomitantly, still larger interstage liquid flow until the point is reached wherein the pressure drop experienced by gases exhausting through the body portion 54 equals the pressure; drop of liquids exhausting through slots 37 of discs 34, at which point the liquid level will stabilize. In practice, the liquid level will hover at a point intermediate the vertical extremities of the cap portion 56 and will be subject to only incipient variations which are immediately compensated by variations in interstage pressure differential and flow rate. It will be apparent that by the use of such a level control in the manner disclosed, there is obtained a continuous monitoring and control of the liquid level in each finisher stage independent of other stages, which control is accomplished with the aid of a simplified device.
Whereas the level control just described is more suitable in operations wherein it is desired to maintain a substanial liquid level within a given stage, such as in the lower five stages, it may be desired to carry out a similar method of finisher stage level control wherein the level is allowed to vary gradually over a wider range. Such requirements may better be satisfied by the employment of a modified form of level control as illustrated in the detail at the lower right of the figure, which embodiment is characterized by a cylindrical body portion 54 similar to that shown in the middle detail view as just described. However, in this modification, the body portion has formed therein an elongated, vertically extending metering slot 58, which may be of varying width, though a uniform width is illustrated.
The upper end of this modified body portion is sealed by means of cap 60 and the lower end thereof sealingly engages one of the perforated discs 34, as by means of threaded portion 62. As the liquid level rises along the body portion to cover a progressively larger portion of the metering slot 58, a progressively smaller area for interstage gas discharge is defined, to thereby generate a greater interstage pressure differential. This interplay continues until that point is reached wherein the pressure drop of the gas flowing through the body portion by way of the metering slot equals the pressure drop of the liquid discharging through the slots 37. By varying the location and dimensions of the metering slot 58, one is enabled to control and stabilize the liquid level over a wide range.
An important advantage to be realized from the practice of this technique of finisher stage level control is the ability to obtain accurate regulation, even through monitoring minute pressure variations. This is most appreciated when one considers the fact that, in polyester systems, it is conventional to operate the finisher at a high vacuum, on the order of 1 mm. of mercury. In order to obtain such high vaccum at the finisher discharge, it has been necessary to operate the upper finisher stages at an undesirably lesser vacuum ranging up to 5 to 7 mm. of mercury. By employing this apparatus, one is enabled to operate the entire finisher at substantially uniform pressure in that the pressure drop across successive stages is, except for very minor transitory variations, negligible to the extent that the uppermost stage pressure may exceed the lowermost stage pressure by as little as a small fraction of a millimeter of mercury.
The reaction product of the finisher zone C, in the form of a finished polymer, is removed from the last stage of the finisher by means of any suitable pump-out mechanism 64, such as a viscoelastic gear, or screw pump, the only consideration being that, due to the high vacuum, imposed on the feed side of such pump-out mechanism, it is desirable to convey the finished polymer from the collection chamber or reservoir 66 to the pump inlet area with a minimum pressure drop. In the case of a gear pump, this may be accomplished by arranging the gear pump housing with respect to the collection chamber 66 in such a manner as to allow the gears to rotate directly in the melt pool contained within the collection chamber to thereby render the pump essentially self-feeding, whereby the hot polymer may be delivered from the high vacuum obtaining within the chamber to a point for further processing.
A vacuum source communicates through port 68 at a point intermediate the lowermost stage of the finisher and the pump-out section 64, 66, which vacuum is in serial communication with each of the finisher stages by way of stage level controls 52 and standpipes 32 of the upper section of the finisher, the pressure drop through the finisher preferably being less than 1 mm, of mercury in the case of a polyester system, pump-out preferably being accomplished at a pressure of no more than 1 mm. of mercury. The following is a specific example of the details of process operation using the apparatus just described.
An initial composition comprising 1,620 cc of ethylene glycol, 1,500 grams of dimethyl terephthalate and 0.975 gram of zinc acetyl acetonate was charged from a bomb into a reactor stage of reactor zone A at a rate of 2,800 cc/hour. The hold-up column of each reactor stage was 477 cc, and a! total of 5 stages were employed with a total capacity of 2,385 cc. The feed temperature was maintained at 220°C. and the reactor zone was operated at a pressure of 75 psig and a temperature of from 260° to 275°C. As the reaction proceeded, the methanol evolved was discharged through a top-side vent and the resulting bis-2-hydroxy-ethyl terephthalate monomer issuing from the reactor zone was passed to the evaporator zone B maintained at atmospheric pressure and a temperature of approximately 275°C.
The zone level controls 28, 29 in both the reactor and evaporator zones were set to maintain approximately 3½ inches of liquid. By operation of flash vaporization and falling film excess ethylene glycol was discharged from the evaporator zone and the bis(2-hydroxyethyl) terephthalate with, possibly, minor portions of very low molecular weight polymer, was passed into the finisher zone C maintained at a temperature of approximately 275°C. and a pressure of 1 mm. of mercury, where the polymerization cycle was brought to a conclusion. The finisher zone employed eleven finisher stages, the first five of which were of the stirred--tank type variety equipped with four inch high standpipes 42, which first five stages had a holdup capacity of 463 cc each, or a total of 2,315 cc. The succeeding six finisher stages were of the plug-flow variety 36, such as shown which latter six stages were operated both with and without packing, according to viscosity variations.
WERNER & PFLEIDERER
A process developed by H. Ocker; U.S. Patent 3,553, 171 ; January 5, 1971; assigned to Werner &. Pfleiderer, Germany involves the production of a spinnable composition and is particularly characterized in that pulverized dimethyl terephthalate (DMT) and ethylene glycol are first mixed to form a homogeneous paste (preferably in a screw extruder with meshing screws and having a very short dwell time). Then the paste is melted and heated to its ester-exchange temperature and is agitated at high speed (preferably using twin paddle screws with pilgrim-step or reciprocating-step motions). After the addition of a suitable catalyst, the mixture is introduced to a series of screw conveyor type reactors at a temperature of about 280°C. (536°F.) and at pressures decreasing from about 60 torrs absolute down to from 0.1 to 0.3 torr absolute with the glycol content from 1 to 3% and the viscosity from 100 to 300 poises in the last reactor.
Among various prior art processes for producing a spinnable polyester composition, the latest process features pulverized DMT which is discharged from a hopper into a tube vibrator and thereafter continues in turn to a melting vessel and then to a cascade. Preheated ethylene glycol introduced from proportioning pumps into the cascade. Ester exchanging takes place in the cascade which comprises several cells provided with agitators. Leaving the cascade, the product flows to a series of reactor vessels which are equipped with vacuum pumps, for degassing. Some of these reactors are also furnished with agitators. A screw conveyor discharges the product for further processing. The afore-described plant according to the prior art also includes other vessels, as well as pumps, piping and auxiliaries. The installation for this process is extremely expensive structurally.
The primary disadvantage of the afore-described prior art process and installation (as well as of other known processes and installations for production of a spinnable polyester composition) was that reliable dwell times in the individual process stages could not be maintained; therefore, in many instances the quality of the final product could not be assured. Variations in dwell times and consequent variations in heating in the various process stages resulted in formation of entirely different molecule chains giving rise to considerable variations in the polymeric finished product. Even the application of extensive control measures did not achieve satisfactory results. This process is designed to cope with these various disadvantages. Figure 5 shows an installation suitable for the conduct of this process.
As seen from the drawing, pulverized DMT and liquid ethylene glycol are fed to screw extruder 1 which has meshing screws 14 driven by motor 15 through transmission 17 and twin shafts 17. Pulverized DMT is fed from hopper 11 by means of endÂless conveyor 18 to inlet port 19. A supply of liquid ethylene glycol is housed in reservoir 20 and is delivered via line 12 to inlet port 19 with flow control thereof provided by regulator 21 . After a very short time, DMT and ethylene glycol exit screw extruder 1 as a homogeneous paste and pass by means of pump 8 via line 22 to a melting and heating apparatus shown as vessel 6, It should be understood that the melting and heating apparatus could also be a screw conveyor heat exchanger (hollow screw shaft). Line 22 is provided with heating jacket 23 having steam inlet 24 and steam outlet 25.
In melting and heating vessel 6 the homogeneous paste is heated to its ester exchange temperature so that ester exchange commences with a consequent release of gaseous methanol. Agitator 7 stirs the mixture and is driven from shaft 26 by motor 27. Vessel 6 is also provided with heating jacket 28 having steam inlet 29 and steam outlet 30. Gaseous methanol exits via port 31.
The liquid product is carried from vessel 6 through exit 32 and line 33 to high speed agitator 2 for ester exchange and further heating accompanied by release of more gaseous methanol. Line 33 has pump 34 which is typical of pumps between elements of equipment in this installation in that it meters product to the succeeding element of equipment, in the case of pump 34 high speed agitator 2. Line 33 is further provided with heating jacket 35 having steam inlet 36 and steam outlet 37. High speed agitator 2 comprises preferably a two shaft pair of paddle screws 38 driven by motor 39 and having a pilgrim step (forward and backward) or reciprocating step motion. Inlet ends 40 and outlet ends 41 of paddle screws 38 have conveying pitches while the mixing portions of the paddle screws are mounted tangentially to each other. Gaseous methanol exits high speed agitator 2 via port 42 and steam enters heating jacket 43 via inlet 44 and exits through outlet 45. The product is removed from high speed agitator 2 through line 46 which is provided with pump 47 as well as heating jacket 48 with steam inlet 49 ond steam outlet 50.
FIGURE 5.APPARATUS FOR THE PRODUCTION OF DIRECTLY SPINNABLE POLYETHYLENE TEREPHTHALATE
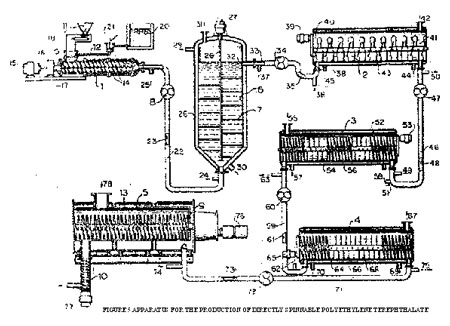
A catalyst such as magnesium, lithium methylate 0.01 to 0.015%, zinc borate 0.2%, lead dioxide, antimony trioxide or cesium oxide is added at 51 and then the still liquid product is introduced to single or twin shaft screw conveyor type reactor 3 in which condensation polymerization begins at approximately 280°C. (536°F.) and about 60 torrs absolute. It is of particular advantage to design first reactor 3 so that screw conveyors 52 driven by motor 53 accurately scrape or strip the product from inside wall 54. Gaseous methanol is vented from port 55 and heating jacket 56 is provided with steam inlet 57 and outlet 58.
The condensation polymerization (or polycondensation) continues in second screw conveyor type reactor 4 at 280°C. and 1 to 10 torrs absolute. The product passes from first reactor 3 to second reactor 4 through line 59 which is provided with pump 60 as well as heating jacket 61 having steam inlet 62 and steam outlet 63. As in first reactor 3 it is of particular advantage to design second reactor 4 so that screw conveyors 64 driven by motor 65 accurately scrape or strip the product from inside wall 66. Gaseous methanol is vented from port 67 and heating jacket 68 is provided with steam inlet 69 and steam outlet 70. With a viscosity of about 100 to 300 poises and an ethylene glycol content of 1 to 3%, the product enters third screw conveyor type reactor 5 for final poly-condensation at 280°C. and 0.1 to 0.3 torr absolute. By selective adjustment from 2,000 to 12,000 poises, the product can be subjected to subsequent treatments such as dyeing, spinning beam, etc. The product is conveyed from second reactor 4 to third reactor 5 by way of line 71 which is provided with pump 72 as well as heating jacket 73 with steam inlet 74 and steam outlet 75. Reactor 5 has twin shaft screw conveyors which are driven by motor 76 in unidirectional rotation and with self-cleansing profiles of engagment. Screw conveyor 10, driven by motor 77, discharges the product for further processing. Vent domes 13 and outlet port 78 are also formed in reactor 5.
An important embodiment of this process in certain cases is to raise the product to from 1,000 to 2,000 poises before it enters third reactor 5 and following this by after polycondensation in reactor 5 at above 8,000 poises.
Prepolymer Production
In the preparation of polyester polymers there is generally first formed a relatively low molecular weight polymer often referred to as "monomer" which has an average degree of polymerization of about 4 or perhaps less. This monomer is conventionally prepared by an ester exchange reaction which comprises condensing a lower dialkyl ester of a dicarboxylic acid or acids with a stoichiometric excess of a glycol or glycols until most of the dialkyl ester has been converted to glycol esters and low polyesters. Such conversion is indicated by the degree of consumption of the theoretically available alkanol.
This condensate which can be called monomer has also been referred to as protopolymer. Neither term however is especially apt. Further heating under conditions facilitating removal of glycol then leads to a gradual increase in molecular weight. At intermediate stages such as at molecular weight ranges of from about 2,000 to about 10,000, the polymer is often spoken of as prepolymer. The intrinsic viscosity of polyethylene terephthalate prepolymers normally ranges from about 0.1 to about 0.55. The final buildup is usually carried to a molecular weight of at least 14,000 to 50,000 or higher. This corresponds to an intrinsic viscosity of 0.5 to 1 .5 or more for polyethylene terephthalate. The degree of polymeri-zation of prepolymers may range from 2 to 15 according to other sources.
REACTOR DESIGN AND OPERATION
Du Pont
A process developed by J. L. Brill and E.F. lzard; U.S. Patent 3,192,184; June 29, 1965; assigned to E.I. du Pont de Nemours and Company involves continuously producing a prepolymer (i .e., a low molecular weight polymer) from bis-2-hydroxy ethyl terephthalate or a low polymer thereof.
In the continuous production of low molecular weight polymers from monomeric substances which polymerize with the evolution of vaporous by-products, such as in the preparation of polyethylene terephthalate from bis-2-hydroxy ethyl terephthalate, the monomer is converted into prepolymer from essentially a melt pool, in stages, viz., a prepolymerization stage and a finishing stage. In the prepolymerization stage the monomer is converted into prepolymer from essentially a melt pool, either in a pot or in a multiple column type of reactor.
This process, conducted under moderate vacuum, takes one hour or more to complete with the result that the partially polymerized material is held up in the apparatus at the severe temperatures necessary for the evacuation of excess vaporous by-products. This material is in the form of a fairly stagnant pool, and consequently, varying amounts of degradation products tend to form. Subsequently, in order to make accept-able films or filaments from the polymer, these degradation products must be removed by means of expensive and often time consuming filtration and/or extraction steps later in the process. The process is designed to minimize polymer degradation and make glycol release rapid and efficient in a continuous process operation.
Briefly stated, the process comprises continuously flowing a liquid polymerizable material from the group consisting of poly-merizable organic monomers and low molecular polymers thereof which polymerize with the evolution of vaporous by-products, said low molecular weight polymers having intrinsic viscosities not greater than 0.25, downwardly and by force of gravity over a substantially vertical surface provided with surface protuberances such as spiral ridges, fins, flutes, grooves or other surface modifi-cation effective to promote distribution of the liquid as a film over the entire surface area, maintaining the film at an elevated temperature and reduced pressure effective to promote progressive polymerization of the film with evolution of vaporous by-product as the film flows down the surface, continuously removing the evolved vaporous byÂproduct in a stream flowing counter-current to the direction of flow of the film, and continuously collecting polymer from said surface at the lower extremity thereof.
An important feature of the process resides in the provision of means for obtaining a continuous i .e., unbroken, film of flowing polymerizable liquid over the substantially vertical surface over which the polymerizabe liquid is caused to flow by gravity. Attempts to flow the liquid over a vertically disposed plane surface were unsuccessful for the reason that the bulk of the liquid tended to form foamy balls which rolled rapidly down the surface in well defined channels, and the material remaining as a film moved sluggishly with the result that polymerization was non-uniform, and only a negligible increase in inherent viscosity could be realized irrespective of the initial viscosity of the liquid feed. However, if the vertical surface is provided with suitable protuberances, as hereinafter described, the surface is satis-factorily wetted and the liquid flows as a continuous film with a substantial absence of channeling or by-passing and progressive polymerization proceeds smoothly to yield a prepolymer of satisfactory inherent viscosity. Figure 1 is a cutaway perspective view of the type of apparatus designed to be used in the process.
As shown in the figure, a plurality of elongated, open-ended tubular reaction columns, each indicated by the reference numeral 1, are vertically disposed in parallel spaced relationship in a closed cylindrical shell or casing 2 which extends substantially beyond the extremities of columns 1. A horizontal feed plate 3 disposed in shell 2 a short distance below the upper extremities of columns 1, and a second horizontal plate 4 located just above the lower extremities of columns 1, define, together with the vertical wall of the casing, an enclosed space surrounding the columns through which space a suitable heating medium may be circulated to maintain the columns at the desired temperature.
The space 5 between plate 4 and the bottom end closure of the shell forms a reservoir for the collection of polymer from the columns. Each of the columns is provided on its inner surface with a continuous spiral protuberance 6, preferably of rectangular cross section, defining a spiral channel down the length of the wall of the column. The protuberance may be integral with the wall of the column, or in the form of a helical ribbon fitted against the wall of the column, or the inner wall of the column may be grooved in a spiral path.
FIGURE 1: CUTAWAY PERSPECTIVE VIEW OF PREPOLYMER PRODUCTION REACTOR
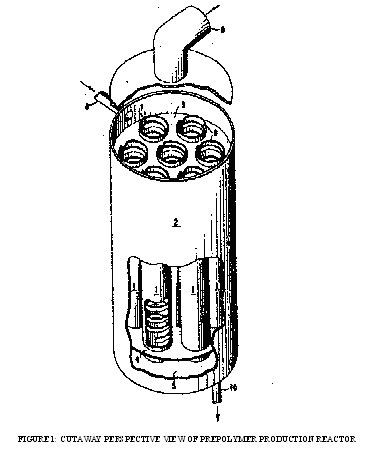
The upper edge of each column is provided with one or more V-shaped weirs or notches 7 adapted to channel polymerizable liquid from the feed plate to the inner wall of the column. A fluid feed inlet conduit 8 with its discharge opening adjacent feed plate 3, and a vacuum duct 9 leading to a conventional vapor take-off and recovery system (not shown) comprising a vapor reflux section and condenser, are located at the upper end of the shell, and an outlet 10 is provided for conducting low polymer from the resevoir at the bottom of the shell to a finisher (not shown) or to storage facilities (not shown).
In operation, the material to be polymerized, e.g., monomeric bis-2-hydroxy ethyl terephthalate or a very low molecular weight polymer thereof (having an intrinsic viscosity of 0.03 to 0.06), is introduced as a continuous stream through conduit B onto feed plate 3 from which it overflows through weirs 7 and by gravity flows downwardly over the inner walls of the columns and into the reservoir 5 below the columns. The liquid being processed does not flow in the channel formed between the spiral protuberances, but rather is forced out of the channel by the formation of gas bubbles formed at the wall of the column and then flows downwardly over a number of coils of the spiral protuberance, re-enters the channel for a short distance, is again forced out and over the protuberance, and so on with the result that the entire wall of the column is wetted with the liquid which flows downwardly as a substantially continuous film.
During the downward flow through the column, the liquid is subjected to temperatures of 240° to 350°C. maintained by cir culation of a suitable heating medium surrounding the columns, and to pressures of 0.1 to 50 millimeters of mercury. Pressure reduction and the removal of vaporous by-products (i .e ., ethylene glycol) is accomplished by application of vacuum through duct 9.
This process may be successfully used either in a two- or three-stage polymerization process for polyethylene terephthalate. It may be utilized in a first stage (flasher) operation to remove 60 to 80% of the excess ethylene glycol in a few minutes time with the resulting product having an intrinsic viscosity of 0.08 to 0.12. Rapid elimination of excess glycol is effected utilizing temperatures of 240° to 280°C. and pressures of 5 to 15 millimeters of mercury.
In a preferred application, the prepolymerization process of the process is carried out in two stages. In this embodiment, up to 85% of the excess glycol is removed from the polymerization system and a prepolymer having an intrinsic viscosity ranging between 0.15 and 0.40 is produced. Polymerization is then completed in a conventional commercial apparatus such as is described in U.S. Patent 2,748,915. The great advantage of this application lies in the fact that the removal of excess ethylene glycol and the preparation of a prepolymer having the desired intrinsic viscosity is carried out in a fraction of the time normally required using conventional prepolymerization processes and apparatus. The total hold-up time of monomer to finished polymer is shortened from an interval of 4 hours to less than 2 ½ hours. The advantages in reduction of expense of operation, increased production and substantial elimination of polymer degradation are readily apparent.
Eastman Kodak
A process developed by K.T. Barkey; U.S. Patent 3,463,760; August 26, 1969; assigned to Eastman Kodak Company involves the preparation of polyester prepolymers for subsequent "solid phase" polymerization (for example, in a heated fluidized bed). It has been found that by using a special method of cooling molten prepolymer, better reactivity in the subsequent solid phase polymerization reaction can be obtained. This special cooling method involves the initial spraying of water droplets under controlled conditions directly onto the top surface of molten prepolymer in order to effect controlled crystallization of the prepolymer at such surface.
In the past, the procedure for cooling the prepolymer melt has usually entailed casting the melt as a fairly thin sheet onto a stainless steel belt, allowing it to cool and subsequently comminuting the particles to appropriate sizes for solid phase polymerization. Such technique is disadvantageous, however, since the total time required to cool a large batch may be as long as two hours or more during which period thermal and oxidative degradations take place resulting in an increase in color in the polyester (which is preferably colorless for most purposes) a decrease in the intrinsic viscosity and an increase in the amount of carboxyl end groups in the polyester.
The latter two effects are undesirable since they increase the time required for solid phase polymerization. Further, it has been found that the higher reaction temperatures required to prepare certain polyester prepolymers, e.g. those polyesters comprising the condensation product of 1, 4-cyclohexanedimethanol and dimethyl terephthalate or the condensation product of dimethyl terephthalate and ethylene glycol, made it difficult to cast them because of rapid coloration even when the melt discharging from the reactor was blanketed with an inert gas such as nitrogen. It was difficult to prevent oxidative degradation and color formation of the polyester at elevated temperatures. Additionally, it has hitherto been necessary to cool prepolymer melts of copolyesters to just above their freezing points before casting onto a stainless steel belt. If the copolyester melt were cooled rapidly, the resultant prepolymer was highly amorphous and too tough to grind.
One method that was suggested heretofore to overcome the problems just described was to "quench" the molten polyester prepolymer in a bath of water immediately after it was poured from the reactor. This was not a satisfactory solution to the problem because such quenching resulted in the formation of a practically completely amorphous prepolymer mass that was also too tough to grind. Evidently, some method, was needed whereby the prepolymer "melt" could be solidified into solid prepolymer, at least a significant proportion (i.e., at least about one-fourth) of which is in the crystalline (rather than amorphous) state.
Accordingly, it is on object of the process to provide a method for cooling a polyester prepolymer in a carefully controlled manner whereby oxidative degradation, discoloration, carboxyl formation and decreased intrinsic viscosity are minimized and the resulting, solidified prepolymer can readily be ground.
In accordance with this process, it has now been found that polyester prepolymer melts (such as, for example, comprising the condensation product of the reaction of 1, 4-cyclohexanedimethanol with dimethyl terephthalate or the condensation product of the reaction of dimethyl terephthalate with ethylene glycol) may be rapidly and efficiently cooled with consequent minimum degradation to form readily grindable, solidified products by the process which comprises the steps of (a) rapidly discharging the melt directly from the prepolymer reactor into a receptacle to form a depth of polyester 0.5 to 30 centimeters thick and preferably 10 to 20 centimeters thick, (b) very shortly thereafter spraying the melt with a fine mist of water, and (c) subsequently flooding the resulting, partially cooled mass of prepolymer with a coolant, preferably water, after a relatively smooth, relatively crack-free layer of at least partially crystallized prepolymer has been formed on the surface of the cooling prepolymer mass.
Apparently, the benefits that can result can be obtained only if certain critical factors are observed during the crucial initial cooling stage of these processes, just after the molten prepolymer has been removed from the reactor. Thus, at this point, before the temperature of the surface of the mass of molten prepolymer has been cooled to below the melting point of the particular prepolymer being prepared, and preferably as soon as the molten material has been discharged (usually under pressure to ensure rapid removal) from the reactor, a spray of water droplets is directed onto the surface of the prepolymer "melt."
It has been found that the size of the water droplets must be maintained below about 300 microns. Preferably, the average diameter of these water droplets should be between about 0.1 and about 200 microns. When only such very small water droplets are sprayed onto the prepolymer "melt," several specific things occur concurrently.
(1) The evaporation of the droplets from the surface of the prepolymer "melt" causes the temperature at the surface of the mass of prepolymer to drop fairly quickly (but not as quickly as it would have if the "melt" had been simply "quenched" into a pool of water).
(2) On the surface of the "melt" there is fairly quickly formed an opaque "scum, " because some of the cooled, solidified polymer is being crystallized.
(3) Although the surface of the melt has solidified, the interior of the mass of cooling prepolymer remains liquid for a certain period of time. This results from the relatively slow transfer of heat from the central portions of the solidifying mass through the solidified outer surfaces, and hence away from the cooling mass.
(4) The opaque surface of the solidifying "melt" remains relatively smooth and relatively crack-free . This is in contrast to that of cooled prepolymer "melt" 0.5 to 12 inches thick that is either air-cooled (or otherwise gas cooled); or immediate "quenching" with coolant as described heretofore which produces an amorphous surface layer that is extremely tough and difficult to break and comminute.
It is the careful control of the water droplet size that makes it possible for all of these events to be accomplished simultaneously to produce the desired results. Thus, if a stream of water (or a spray of water droplets having average diameters above about 500 microns for example) were directed onto the surface of the "melt," the surface would immediately be "churned" into a foam or froth (due to temporarily entrained water being explosively changed to steam), and the temperature of the mass of cooling prepolymer would be lowered so quickly that the resulting cooled material would be too highly amorphous to grind readily.
Note that in the processes, the rate of cooling of the overall prepolymer mass is effectively "automatically" controlled within the range desired to produce the necessary, partially crystallized, prepolymer when the several specified parameters set out herein are carefully followed. For example, not only must the water droplet size be carefully controlled, but also the total amount of water that is sprayed onto the surface of the molten mass must be carefully regulated during the critical first few minutes of the water spray treatment.
Thus, during the period in which the layer of relatively smooth, partially crystallized, solidified polymer "scum" is being formed on the surface of the "melt" (the total time depending upon several factors including initial temperature, and water spray rate) between about 0.1 and about 10 pounds (and preferably between about 0.5 pound and about 4 pounds) of water per 1,000 square feet of prepolymer "melt" surface per minute should be sprayed onto the surface.
This rate should be maintained until the temperature of the surface being sprayed is lowered to below about 50°C. less than the melting point of the prepolymer. Preferably, the rate of water spray should be maintained so that between about 5 seconds and about 5 minutes are required for the temperature of the outer surface of the prepolymer mass to pass from about 230° to about 140°C.
The foregoing rate data are described in terms of pounds of water per minute per 1,000 square feet of prepolymer surface area because the processes are not dependent upon the actual amount of surface (or conversely, thickness of the layer of prepolymer "melt") so long as the layer is at least about 0.5 centimeter thick. For this reason, the particular shape of the container into which the prepolymer "melt" is dumped or poured from the reactor is not significant, insofar as the successful practice of this process is concerned.
Thus, the "melt" can be water-spray treated on a moving belt, in a "batch" type container such as an ingot mold, in a trough or series of troughs on a conveyor system, or any of a number of ways readily appreciated by the skilled artisan. If desired, the tiny water droplets described above can contain in admixture therewith any of a number of readily volatilizable materials (such as alcohols, ketones, such as acetone, alkylene halides, alkyl halides, and the like) having boiling points below about 140°C. in amounts up to about 50 weight percent, but preferably less than 25 weight percent, of the water contained therein.
One other factor that is also essential to the successful practice of this process is the flooding of the cooling prepolymer mass with a coolant after the desired, relatively smooth, partially crystallized "scum" layer has been formed, but before the entire mass of prepolymer has been solidified. This coolant can be in the form of relatively pure water, or practically any other material that is liquid at room temperature (such as ethanol, ethyl ether, methanol, acetone, isopropyl alcohol, methyl ethyl ketone, fluorinated hydrocarbons, and the like) and that can be readily separated from the resulting solidified prepolymer product when the separation is desired. It can also be a mixture of water with a water miscible volatile solvent such as methanol, ethanol or acetone, or even a volatile material alone, such as methylene chloride, the aforeÂmentioned materials, and the like; although it is preferably water.
The "flooding" can be accomplished gradually (as by continuing the spraying described above until liquid water remains in contact with the surface of the prepolymer) or suddenly (as by dunking the prepolymer into a cool water bath). That such "flooding" is essential, however, is evident from the observation that, if the spray of water droplets is halted at the time the desired "scum" is formed, but before the surface temperature of the cooling prepolymer mass drops to 100°C. and no "flooding" is undertaken, either the "scum" layer will disappear or its beneficial effects will disappear due to the sharply increased surface temperatures that are caused by the large amount of heat still remaining in the central portions of the cooling prepolymer mass. Thus, "flooding" is essential, and should be maintained at least until the entire prepolymer mass has solidified, and preferably until the temperature of the central portion of the mass drops below about 200°C.
Inventa AG
An apparatus developed by.W.-D. Hahn; U.S. Patent 3,544,522; December 1, 1970; assigned to Inventa AG fur Forschung and Patentverwertung, Switzerland is useful for the conduct of various stages of polyester manufacture, including prepolymer production. Figure 2 shows a sectional view of the apparatus.
The apparatus includes a rotably mounted, horizontally extending drum 2 have an internal continuous helical baffle 10 extending inwardly from the internal surface of the drum 2. The baffle at its outer periphery is fixedly connected with the drum, as by welding. Further, the baffle terminates short of the center of the drum.
Thereby, the successive turns of the baffle are communicated by openings 15 defined by the inner periphery of the baffle. The drum is mounted on trunnions 5, 5' and is outfitted with a centrally disposed feed pipe 1 . Gearing 8 is connected to the drum at the inlet end and is also connected, through a variable speed transmission 7, to drive motor 6. Accordingly, the drum can be rotated at different rates. Opposite the inlet end, the drum is provided with an opening 18 for discharge of material 11 which is advanced through the drum by rotation thereof.
A vessel 3 encloses and hermetically seals the drum, and is outfitted with conduit means 13 for communicating the vessel with a pressure bearing device, for example the vacuum pump 19. Further, means are provided, for communicating the interior of the drum 2 and the vessel for communicating successive turns of the baffle 10 with the vessel 3 and therethrough with the pressure bearing device. Thus, the inlet end of the drum is provided with an opening 14 and, the outlet end of drum is provided with an opening 16, each of these openings communicating the inside of the drum with the inside of the vessel 3. Further, the openings between successive turns of the helical baffle 10, which are defined by the inner periphery 15 of the baffle, communicate the space between successive turns of the baffle with the openings 14 and 16 in the ends of the drum. The apparatus is also provided with means for heating or cooling the drum. Thus, the apparatus is provided with a jacket 9 through which a heating or cooling medium can be passed. A conduit system 20 comprising an outer pipe 21 and an inner pipe 22 passes into the drum from the outlet end thereof. Within the drum is a pivot fitting 23 connected on its inlet side to the outer pipe 21 for forward communication therewith and connected on its outlet side to conduit 24 which is communicated with the inside of the jacket 9.
FIGURE 2: HORIZONTAL REACTOR DESIGN SUITABLE FOR POLYESTER PREPOLYMER PRODUCTION

The conduit 24 turns with the drum, the pivot fitting 23 permitting this. The inner pipe 22 passes through the pivot fitting 23 and onto the pivot fitting 25. The pivot fitting 25 has its inlet side connected to the pipe 22 and its outlet side connected to the conduit 26 which communicates with the inside of the jacket 9. The conduit 26 turns with the drum, the pivot fitting 25 permitting such movement. Heating or cooling fluid can be passed through the jacket utilizing the pipe in pipe system 20.
The following is a specific example of the use of such an apparatus in the manufacture of polyesters prepolymer. 41 .0 kg. of bis-(2-hydroxethyl) terephthalate with a condensation polymerization degree of about 2 and a viscosity of 4 cp. was heated to a temperature of 230° to 350°C. and fed to the drum at 1 The drum revolved at a speed of 8 revolutions per hour. A vacuum of about 180 torr was applied to the drum. The temperature of the reaction mixture rose during the time of stay of 1 .5 hours to 260° to 270°C. and the viscosity rose to about 40 p. The end product had an intrinsic viscosity of 0.15 according to a measurement made in a mixture of 60 weight percent phenol and 40 weight percent 1,1,2,2- tetra-chlorethane. The same type of device, it should be noted, can be used in the manufacture of DMT from TPA, in the manufacture of BHET and also in the final polymerization (finishing) step in polyester manufacture.
